Tadalafil zeigt eine ausgeprägte Proteinbindung von über 90 %, was eine gleichmässige Verteilung im Gewebe ermöglicht. Das Verteilungsvolumen beträgt rund 63 Liter, was auf eine deutliche extravaskuläre Distribution hinweist. Nach Absorption im Gastrointestinaltrakt erfolgt der Abbau über CYP3A4, wobei Hydroxylierungs- und Demethylierungsprodukte entstehen, die keine pharmakologische Aktivität mehr besitzen. Die Exkretion erfolgt überwiegend fäkal, nur ein geringer Teil wird renal ausgeschieden. Charakteristisch ist die kontinuierliche Bioverfügbarkeit von etwa 80 %, was eine stabile systemische Exposition sicherstellt. Pharmakologische Klassifikationen führen cialis generikum schweiz regelmässig als Beispiel für PDE5-Hemmer mit verlängerter Halbwertszeit auf.
Pq140008151p
Design of a potent and selective inhibitor of the intermediate-conductance Ca2؉-activated K؉ channel, IKCa1: A potential immunosuppressant Heike Wulff*†, Mark J. Miller*, Wolfram Ha¨nsel‡, Stephan Grissmer§, Michael D. Cahalan*, and K. George Chandy*
*Department of Physiology and Biophysics, University of California, Irvine, CA 92697; ‡Pharmaceutical Institute, University of Kiel, 24118 Kiel, Germany;and §Department of Applied Physiology, University of Ulm, 89081 Ulm, Germany
Edited by Lily Y. Jan, University of California, San Francisco, CA, and approved May 15, 2000 (received for review March 14, 2000)
The antimycotic clotrimazole, a potent inhibitor of the intermedi-
activation response (19). In contrast, mitogen-activated human
ate-conductance calcium-activated K؉ channel, IKCa1, is in clinical
T lymphocytes exhibit 300–800 functional IKCa1 channels (20)
trials for the treatment of sickle cell disease and diarrhea and is
along with 400–500 Kv1.3 channels. Because expression of
effective in ameliorating the symptoms of rheumatoid arthritis. IKCa1 channels is dramatically enhanced in activated T cells
However, inhibition of cytochrome P450 enzymes by clotrimazole
(20), in parallel with enhanced [Ca2ϩ]i signaling (21, 22), a
limits its therapeutic value. We have used a rational design strat-
strategy targeting IKCa1 channels could be especially effective in
egy to develop a clotrimazole analog that selectively inhibits IKCa1
suppressing chronically activated T cells and could perhaps lead
without blocking cytochrome P450 enzymes. A screen of 83 tri-
to therapy for autoimmune disorders. arylmethanes revealed the pharmacophore for channel block to be
By identifying and exploiting differences in the pharmacoph-
different from that required for cytochrome P450 inhibition. The
ores required for channel block and cytochrome P450 inhibition,
‘‘IKCa1-pharmacophore’’ consists of a (2-halogenophenyl)diphe-
we have designed a triarylmethane (TRAM-34) that selectively
nylmethane moiety substituted by an unsubstituted polar -elec-
blocks the IKCa1 channel. TRAM-34 may have a therapeutic
tron-rich heterocycle (pyrazole or tetrazole) or a ؊C'N group,
profile similar to clotrimazole but may lack its toxic side effects. whereas cytochrome P450 inhibition absolutely requires the imi- dazole ring. A series of pyrazoles, acetonitriles, and tetrazoles Materials and Methods were synthesized and found to selectively block IKCa1. TRAM-34 Compounds. Clotrimazole (1a), econazole, and ketoconazole (1-[(2-chlorophenyl)diphenylmethyl]-1H-pyrazole) inhibits
were purchased from Sigma. Clotrimazole was subjected to the
cloned and the native IKCa1 channel in human T lymphocytes with
same physical analysis as the synthesized triarylmethanes (see
a Kd of 20 –25 nM and is 200- to 1,500-fold selective over other ion
supplementary Table 2, www.pnas.org) to ensure its purity. channels. Using TRAM-34, we show that blocking IKCa1 in human
Nifedipine, nimodipine, and nitrendipine were obtained from
lymphocytes, in the absence of P450-inhibition, results in suppres-
Research Biochemicals. Triarylmethanes were synthesized ac-
sion of mitogen-stimulated [3H]thymidine incorporation of preac-
cording to the route described for clotrimazole (23) with mod-
tivated lymphocytes with EC50-values of 100 nM-1 M depending
ifications according to ref. 24 and of our own. Compounds were
on the donor. Combinations of TRAM-34 and cyclosporin A are
characterized by melting point, IR, 1H-NMR, mass spectrome-
more effective in suppressing lymphocyte mitogenesis than either
try, and combustion analysis. Briefly, triarylmethanols (2a-p) compound alone. Our studies suggest that TRAM-34 and related
were prepared from benzophenones and aryl bromides by a
compounds may hold therapeutic promise as immunosuppressants.
Grignard reaction and then converted into triaryl chlorides with
freshly distilled thionyl chloride in petroleum ether, which then
Clotrimazole,atopicallyusedantimycotic,exertsitsfungicidal were further reacted with an excess of the required amine in
effect by inhibiting fungal P450-dependent enzymes (1).
anhydrous acetonitrile to give compounds 1b-f, 3h-l, 4a-q, 6a-m,
Clotrimazole has also been reported to inhibit mammalian P450
and 7a. Bivalent and trivalent compounds 8a-f were synthesized
enzymes (2–4), as well as directly block the intermediate-
according to ref. 25. Compounds 3a-d were prepared from the
conductance Ca2ϩ-activated potassium (IKCa) channel, a prod-
triaryl chlorides in a mixture of diethyl ether and 25% aqueous
uct of the IKCa1 gene (5–7), in human erythrocytes, colonic
ammonia solution (26). Compounds 3e-g were prepared by
epithelium, and human T lymphocytes at nanomolar concentra-
reacting 3a, 3b, and 3d with freshly distilled acetic anhydride.
tions (8–13). This lack of specificity clouds the use of clotrim-
Compounds 3h-k were synthesized from triaryl chlorides and
azole as a pharmacological tool, creating a need for a truly
urea according to the method given for 8a-f. Compounds 5a-f
selective IKCa1 inhibitor. Because of its potent channel-blocking
were synthesized by heating triaryl chlorides with copper cyanide
activity, clotrimazole is being clinically evaluated for the treat-
ment of erythrocyte dehydration in sickle cell disease and
secretory diarrheas (9, 14). Recent studies have also raised the
Clones, Cells, and Cell Lines. The cloning of human IKCa1 and
possibility of using clotrimazole as an immunosuppressant (12,
transient transfection into COS-7 cells have been previously
13). Clotrimazole was previously reported to be effective in
rheumatoid arthritis (15). However, the gastrointestinal and
urinary disturbances caused by clotrimazole, coupled with ele-
This paper was submitted directly (Track II) to the PNAS office.
vation of hepatic enzymes (9, 16) and changes in plasma cortisol
Abbreviations: CRAC, calcium release-activated Ca2ϩ; KCa, Ca2ϩ-activated Kϩ; IKCa, inter-
levels (15) caused by its acute inhibition (2–4) and chronic
mediate-conductance KCa; PBMCs, peripheral blood mononuclear cells; PMA, phorbol-12-
induction of human P450-dependent enzymes (3, 17), may limit
myristate 13-acetate; [3H]TdR, tritiated thymidine.
†To whom reprint requests should be addressed at: Department of Physiology and Bio-
Resting human T lymphocytes possess Ϸ400 Kv1.3 channels
physics, University of California Medical School, Joan Irvine Smith Hall, Room 291, Irvine,
and roughly 2–20 functional IKCa1 channels. The membrane
potential of resting T cells is maintained by Kv1.3 channels rather
The publication costs of this article were defrayed in part by page charge payment. Thisarticle must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. PHARMACOLOGY
than by IKCa1, and selective inhibitors of Kv1.3 suppress the
§1734 solely to indicate this fact.
PNAS ͉ July 5, 2000 ͉ vol. 97 ͉ no. 14 ͉ 8151– 8156
Inhibitory effects of compounds 1a (clotrimazole), 1c, and 5a on IKCa1 currents expressed in COS-7 cells. Voltage ramps were applied from Ϫ120 mV
reported (11). Cell lines stably expressing mKv1.1, rKv1.2,
Inhibition Studies of CYP3A4. Inhibition of the catalytic activity of mKv1.3, mKv3.1, and hKv1.5 have been previously described
purified recombinant human cytochrome P450 3A4 in micro-
(28). Human SKCa2 (expressed sequence tag: GenBank acces-
somes (Gentest Corporation, Woburn, MA) was assayed on the
sion no. AI810558) and human SKCa3 (AJ251016), were cloned
turnover of 7-benzyloxy-4-trifluoromethyl-coumarin by the de-
in-frame downstream to green fluorescent protein in the
tection of its fluorescent metabolite 7-hydroxy-4-trifluorometh-
pEGFP-C1 expression vector (CLONTECH). Rat SKCa2
ylcoumarin as described (33). All experiments were done in
(U69882) was previously described (29). These clones were
duplicate, and results are reported as percent inhibition. Positive
transiently expressed in COS-7 cells. LTK cells expressing hKv1.4
controls (5 M ketoconazole and 100 nM clotrimazole) were run
and rKv4.2 were obtained from M. Tamkun (University of
on the same plate producing 99% inhibition.
Colorado, Boulder, CO), HEK-293 cells expressing the skeletal
muscle sodium channel hSkM1 (SCN4A) from F. Lehmann-
[3H]Thymidine Incorporation Assay. Resting or 2-day-activated (10
Horn (University of Ulm, Ulm, Germany), and HEK-293 cells
nM PMA or 5 ng͞ml anti-CD3 Ab) PBMCs were seeded at 2 ϫ
expressing hSlo␣ (30) from A. Tinker (Centre for Clinical
105 cells per well in culture medium in flat-bottom 96-well plates
Pharmacology, University College London). Peripheral blood
(final volume 200 l). Cells preincubated with drug (60 min)
mononuclear cells (PBMCs) were isolated from heparinized
were stimulated with mitogen (10 nM PMA ϩ 175 nM ionomycin
blood samples of healthy volunteers by using a lymphocyte
or 5 ng͞ml anti-CD3 Ab) for 48 h. Triated thymidine ([3H]TdR)
separation medium (Accuspin System-Histopaque-1077, Sigma)
(1 Ci per well) was added for the last 6 h. Cells were harvested
and maintained in RPMI-1640 supplemented with 10% FCS͞2
onto glass fiber filters and radioactivity measured in a scintilla-
mM L-glutamine͞1 mM Naϩ pyruvate͞1% nonessential amino
acids͞100 units/ml penicillin͞100 g/ml streptomycin. Cells were
rested for 24 h after isolation and then activated with 10 nM
Flow Cytometric Measurement of Cell Viability. Cells were seeded at
phorbol-12-myristate 13-acetate (PMA) or 5 ng/ml anti-CD3 Ab
5 ϫ 105 cells͞ml (Jurkat E6–1, MEL cells, human T lympho-
(Biomedia, Foster City, CA). T cells were isolated by nylon-wool
cytes) or 105 cells͞ml (C2F3 myoblasts, CHO, COS-7, L929, NGP
purification immediately before electrophysiological experi-
and NLF neuroblastoma, RBL-2H3) in 12-well plates. Drug (5
ments, typically yielding Ͼ90% CD3ϩ T cells.
M) was added in a final DMSO concentration of 0.1% which
was found not to affect cell viability. After 48 h cells were
harvested by suction (suspension cells) or by trypsinization
Electrophysiology. Cells were studied in the whole-cell configu-
ration of the patch-clamp technique. The holding potential in all
(adherent cell lines), centrifuged, resuspended in 0.5 ml PBS
experiments was Ϫ80 mV. For measurement of IK
containing 1 g͞ml propidium iodide (PI), and red fluorescence
measured on a FACScan flow cytometer (Becton Dickinson).
BKCa currents, we used an internal pipette solution containing
The percentage of dead cells was determined by their PI uptake,
(in mM): 145 Kϩ aspartate, 2 MgCl2, 10 Hepes, 10 K2 EGTA, and
104 cells of every sample being analyzed.
8.5 CaCl2 (1 M free Ca2ϩ), pH 7.2, 290–310 mOsm. To reduce
currents from the native chloride channels in COS-7, T84, and
Acute in Vivo Toxicity Determinations. Five CF-1BR mice (17–19 g)
T cells, Naϩ aspartate Ringer was used as an external solution
were injected intravenously with a single 1.0-ml dose of 0.5
(in mM): 160 Naϩ aspartate͞4.5 KCl͞2 CaCl2͞1 MgCl2͞5 Hepes,
mg͞kg TRAM-34 (in mammalian Ringer solution with 1%
pH 7.4͞290–310 mOsm. IKCa currents in COS-7 and T84 cells
ethanol and 2.5% BSA). Five control mice were injected with an
were elicited by 200-ms voltage ramps from Ϫ120 mV to 40 mV
equal volume of the vehicle. Mice were observed for adverse
applied every 10 s and the reduction of slope conductance at Ϫ80
effects immediately after dosing, at 4 h after injection and daily
mV by drug taken as a measure of channel block. For activated
T lymphocytes, the same voltage ramp was applied every 30 s to
avoid inactivation of Kv1.3 channels. BKCa currents were elicited
by 200-ms voltage ramps from Ϫ80 to 80 mV applied every 30 s
Defining the Triarylmethane Oharmacophore for IKCa1 Block. Fig. 1
and channel block measured at 35 mV. The inward rectifier
shows currents from IKCa1-transfected COS-7 cells elicited by
(rKir2.1) in RBL cells was studied in Naϩ aspartate Ringer with
voltage ramps with 1 M free calcium in the pipette solution.
a Kϩ aspartate-based pipette solution containing 50 nM free
Clotrimazole (compound 1a) potently blocks the IKCa1 channel
Ca2ϩ. Recordings from Jurkat SKCa channels were made in Kϩ
with a Kd of 70 nM. In contrast, two related antimycotic agents,
aspartate Ringer. For both SKCa and inward rectifier currents,
ketoconazole (Kd ϭ 30 M) and econazole (Kd ϭ 12 M), as well
the reduction of slope conductance at Ϫ110 mV was taken as
as the dihydropyridines nifedipine (Kd ϭ 4 M), nimodipine (Kd
measure of channel block. For all currents elicited by voltage
ϭ 1 M), and nitrendipine (Kd ϭ 0.9 M), are significantly less
ramps, series resistance was not used. Recordings of Kv- (28),
monovalent currents through Jurkat calcium release-activated
We synthesized 83 triarylmethanes and tested them by whole-
Ca2ϩ (CRAC) channels (31), and swelling-activated chloride
cell patch clamp against IKCa1 channels, the compounds being
currents (32) were made as previously described.
added externally in every case. The structures and channel-
8152 ͉ www.pnas.org
blocking potencies of 30 exemplary compounds that highlight
our design strategy are described in Fig. 2, and their physical data
are listed in supplementary Table 3 (www.pnas.org). The struc-
tures and channel affinities of the remaining 53 compounds are
provided in supplementary Table 3 and their physical data in
supplementary Table 4. The hydrolytic stability of TRAM-34 is
To test whether the imidazole ring is necessary for channel
blocking activity, we generated several analogs where this moiety
is replaced by a hydroxyl- (2a-p), an amino- (3a-d), an acet-
amido- (3e-g), an ureido- (3h-k), a malono- (3 l), an aromatic
pyrrole- (4a), an aminothiazol- (4d), or an aminopyridine- (4k)
group. All these analogs are significantly less potent than
clotrimazole (Fig. 2 A), indicating the need of the imidazole
moiety for channel block. Five bivalent compounds and one
trivalent compound (supplementary Table 3, 8a-f ) are inert. The
triphenylmethyl moiety of the molecule is equally important for
channel block, because replacement of one or more of the phenyl
rings by thiophene (1c, 2 m-p) or pyrimidine (supplementary
Table 3) reduces activity 10- to 20-fold (Fig. 2 A). Our analysis
also reveals the requirement of the o-halogen on the triphenyl-
methane, because imidazole analogs lacking an o-chlorine sub-
stituent (1b) are 20-fold less potent than clotrimazole (1a),
whereas compounds containing more than one chlorine (2d-e,
supplementary Table 3) are inert. Collectively, our data indicate
that low nanomolar block of IKCa1 requires the presence of
both the (2-halogenophenyl)diphenylmethane and the imidazole
Comparison of the Pharmacophores for Channel Block and Cyto- chrome P450 Inhibition. Extensive structure-activity studies of
azole antimycotics have shown that the imidazole ring is abso-
lutely required for block of cytochrome P450 enzymes. These
compounds exert their inhibitory effect by coordinately binding
to the heme iron of P450-dependent enzymes with the N3
nitrogen of the imidazole ring (1). Replacement of the imidazole
ring by other heterocyles lacking this crucial nitrogen atom
abolishes inhibition and induction of cytochrome P450 enzyme
activity (17, 23). To determine whether such substituents might
retain potency against IKCa1, we generated a new series of
analogs (Fig. 2B) where the imidazole moiety was replaced with
functional groups of similar size, lipophilicity, and -electron
density, such as ϪC'N (5a-d), pyrazole (6a-m), and tetrazole
(7a). Two acetonitriles (5a, 5b), four pyrazoles (6b-e), and the
tetrazole (7a) analog are potent inhibitors of IKCa1, four of these
having higher affinities than clotrimazole (Fig. 2B). However,
any substitution on these small heterocycles (6h-k) dramatically
diminishes affinity (Kd 1–25 M), an effect we also witnessed in
the corresponding imidazole compounds (Fig. 1, Table 1, 1c, 1e,
and 1f, and supplementary Table 3, www.pnas.org). As with the
imidazole series, the o-halogen is required for optimal activity,
because 6a lacking this group is 100-fold less effective. Ten
compounds, including representative acetonitriles (5a, 5b) and
pyrazoles (6b, 6k), were tested at a single high concentration of
10 M for inhibition of the catalytic activity of recombinant
human cytochrome P450 3A4, the major xenobiotic metabolizing
Kd values determined by fitting the data with the same Hill coefficient as clotrimazole. Inhibition of CYP3A4 was tested at 10 M except for clotrim- azole, which was tested at 100 nM. (B) Structures of triarylmethyl- acetonitriles, -pyrazoles, and -tetrazoles, and blocking potencies of IKCa1 and CYP3A4. Compound 6b was tested at five concentrations (15 cells); the other
(A) Structures of triarylmethanes and blocking potencies of IKCa1 and
compounds were tested at three concentrations (9 cells). Hill coefficient ϭ 1.0
CYP3A4. Clotrimazole and compound 1a were tested at five concentrations
to 1.2. (C) Pharmacophore for triphenylmethane IKCa1 blockers. (Left) AM1-
(n ϭ 3). Kd and Hill coefficient (Hill coefficient ϭ 1.2) were determined by
optimized molecular structure of TRAM-34 (color code: white, hydrogen and
fitting the Hill equation to the reduction of slope conductance at Ϫ80 mV. The
chlorine; light gray, carbon; black, nitrogen). (Right) General structure of the
PHARMACOLOGY
remaining compounds were screened at 100 nM, 1 M, and 10 M and their
PNAS ͉ July 5, 2000 ͉ vol. 97 ͉ no. 14 ͉ 8153 Table 1. Selectivity TRAM-34 Is a Highly Selective Inhibitor of Cloned IKCa1 and Native IKCa Currents. A pyrazole 1-[(2-chlorophenyl)diphenylmethyl]-1H.
pyrazole (6b, TRAM-34) was characterized further. This highly
lipophilic compound (logP value of 4.0) is readily membrane
permeable. Fig. 3A shows the effect of externally applied
TRAM-34 on IKCa1 currents in COS-7 cells. The dose-response
curve (Fig. 3B) reveals a K
of 1.2 with 1 M calcium in the pipette. Because the IKCa1
channel is activated by cytoplasmic calcium (half activation:
Ϸ300 nM) via a calmodulin-dependent mechanism and is not
voltage dependent (7, 11, 20), we examined whether the chan-
nel’s sensitivity to block by TRAM-34 depends on the intracel-
lular calcium concentration. The Kds measured at lower internal
calcium concentrations (500 nM Ca2ϩ Kd ϭ 24 Ϯ 8 nM; 250 nM
Ca2ϩ Kd ϭ 28 Ϯ 6 nM) suggest that block by TRAM-34 is not
calcium dependent. The block by all triarylmethanes is voltage
independent and slow in onset, taking 3–6 minutes to reach
Activation of human T lymphocytes via the receptor signaling
complex by anti-CD3 Ab or the PKC-dependent cascade by
PMA results in a 20- to 50-fold increase in IKCa1 conductance
after 48 h (Fig. 3C). Currents at potentials more negative than
Ϫ40 mV are through the IKCa1 channel, whereas at more
depolarized potentials, Kϩ currents are carried by a combination
of IKCa1 and the voltage-gated Kϩ channel, Kv1.3. As shown in
Fig. 3D, TRAM-34 selectively blocks the IKCa1 current (Kd ϭ
enzyme in human liver. These compounds do not inhibit
25 nM), whereas the residual Kv1.3 current is blocked by the
CYP3A4 activity at 10 M, whereas clotrimazole, for which
selective peptide inhibitor, ShK-Dap22 (22, 36). TRAM-34 also
blocks IKCa1 currents in human T84 colonic epithelial cells with
50 values vary from 250 pM (34) to 30 nM (4),
completely inhibits CYP3A4 at 100 nM (Fig. 2). Thus, we have
To test the selectivity of the compound, we screened it against
successfully separated the IKCa1-blocking activities from cyto-
a panel of 15 other channels (Table 1). TRAM-34 is 200- to
1,500-fold less effective against several related mammalian po-
Our results suggest that optimal potency against the IKCa1
tassium channels: Kv1.1-Kv1.5, Kv3.1, Kv4.2, Kir2.1, BK
channel is achieved with a (2-halogenophenyl)diphenylmethane
moiety substituted by a small unsubstituted polar -electron-rich
hSKCa2, hSKCa3) as well as the native SKCa in Jurkat T cells.
heterocyle (pyrazole or tetrazole) or a ϪC'N group (Fig. 2C).
The CRAC calcium channel, the human SKMI-sodium channel,
Molecular modeling studies (AM1) render a propeller-shaped
the swelling-activated chloride channel in activated human T
structure for the pharmacophore. The three phenyl rings are
lymphocytes, and the native chloride channel in COS-7 cells are
almost perpendicular to the central CON bond axis between the
triphenylmethane moiety and the imidazole or pyrazole ring.
This modeled structure is in agreement with the crystal structure
TRAM-34 Suppresses Human T Lymphocyte Activation. Jensen et al.
(13) recently showed that 10 M clotrimazole suppresses anti-
(A) IKCa1 currents in COS-7 cells blocked by TRAM-34. (B) Dose response for IKCa1 channel block by TRAM-34. The Hill equation was fitted to the
reduction of slope conductance at Ϫ80 mV (15 cells). (C) IKCa currents in resting human T lymphocytes and in T lymphocytes activated for 2 days with PMA oranti-CD3 Ab. Mean IKCa conductance in resting cells ϭ 0.098 (Ϯ0.17) ns (n ϭ 24), PMA activated (10 nM) ϭ 3.45 (Ϯ2.21) ns (n ϭ 21), anti-CD3 Ab-activated(5 ng͞ml) ϭ 5.59 (Ϯ3.91) ns (n ϭ 20). (D) Effect of TRAM-34 and ShK-Dap22 on Kϩ currents in activated T lymphocytes. 8154 ͉ www.pnas.org
gen- and mitogen-induced proliferation of resting human lym-
phocytes. Since this concentration is Ϸ100 times the channel-
blocking dose, suppression is probably due to a nonspecific
mechanism. Studies done at the same time by Khanna et al. (12)
showed that 250 nM clotrimazole (a concentration closer to the
channel-blocking dose) suppresses the activation of phytohe-
magglutinin-preactivated T cells more effectively than the acti-
vation of quiescent cells. However, because clotrimazole blocks
both IKCa1 and cytochrome P450 enzymes, the mechanism
underlying this suppression remains unclear.
We have used TRAM-34 to evaluate the role of IKCa1 in
resting and activated lymphocytes. Quiescent cells were activated
for 48 h through the T-cell-receptor signaling pathway with
anti-CD3 Ab or with a combination of the PKC-activator PMA
and calcium-ionophore ionomycin, in the presence or absence of
TRAM-34, and the incorporation of [3H]TdR measured. In
parallel, cells were preactivated with either anti-CD3 Ab or PMA
for 2 days to up-regulate IKCa1 channels and then restimulated
with the mitogenic combinations used on quiescent cells. Up-
regulated IKCa1 expression, to a level of several hundred
channels in T cells preactivated by either stimulus, was con-
firmed in four of the six donors by whole-cell recording (n ϭ
20͞donor). In keeping with our expectations, TRAM-34 sup-
presses reactivation of lymphocytes by both mitogenic stimuli
(Fig. 4 A and B, closed symbols). Sensitivity varies with the
different stimuli and from donor to donor. In anti-CD3 Ab-
stimulated T cells, the mean EC50 value among sensitive donors
is 295 (Ϯ130) nM and 910 (Ϯ70) nM for less sensitive donors. In
PMA ϩ ionomycin-activated cells, including both T and B
lymphocytes, the EC50 values are 85 (Ϯ30) nM for sensitive and
830 (Ϯ300) nM for less sensitive donors. In contrast, TRAM-34
has little effect at nanomolar concentrations on the activation of
resting human lymphocytes and requires a dose 250–500 times
the channel-blocking dose (5–10 M) to inhibit [3H]TdR incor-
poration (Fig. 4 A and B, open symbols), which may be caused
by nonspecific effects. Thus, our results with TRAM-34 dem-
onstrate that selective blockade of IKCa1 channels preferentially
suppresses mitogenesis in preactivated lymphocytes, in response
to either PMA ϩ ionomycin or to specific T-cell stimulation via
TRAM-34 Combined with Cyclosporin A. Cyclosporin A inhibits
T-cell proliferation by acting on the calcineurin-dependent step
in the activation cascade (19), whereas TRAM-34 acts on an
Effect of TRAM-34 on anti-CD3 Ab- (A) or PMA ϩ ionomycin- (B)
earlier event, namely the modulation of calcium entry. A com-
stimulated [(3H)-TdR incorporation by resting and preactivated lymphocytes.
bination of the two compounds might therefore suppress mito-
PBMCs from different donors were activated with anti-CD3 Ab (5 ng͞ml) or a
genesis more substantially than either compound alone. To test
combination of PMA (10 nM) ϩ ionomycin (175 nM) for 48 h. [3H]TdR was
this idea, preactivated T cells were stimulated with PMA and
added to the culture for the last 6 h. In parallel, PBMCs were preactivated with
ionomycin in the presence or absence of cyclosporin A and
either anti-CD3 Ab (5 ng͞ml) or 10 nM PMA for 48 h (to up-regulate IKCa1
increasing doses of TRAM-34. The dose response for cyclo-
channels) and then restimulated for a further 48 h with anti-CD3 Ab or PMAϩ ionomycin. Donor 1 resting (ᮀ), donor 2 resting (E), donor 3 resting (⌬),
sporin A-mediated suppression of [3H]TdR incorporation was
donor 1 preactivated (■), donor 2 preactivated (F), donor 3 preactivated (Œ),
shifted by TRAM-34 to more sensitive values by a factor of 2-
donor 4 preactivated (ࡗ), donor 5 preactivated [X, donor 6 preactivated (Ϫ)].
to 10-fold for donor 1 (Fig. 4C). Similar results were obtained
(C) Effects of TRAM-34 on cyclosporin A-mediated inhibition of [(3H)-TdR
with donors 2 and 6 (data not shown).
incorporation. PBMCs from donor 1 were preactivated with 10 nM PMA for48 h and then restimulated for a further 48 h with PMA ϩ ionomycin in thepresence or absence of cyclosporin A and TRAM-34. [3H]TdR was added to the
TRAM-34 Is Nontoxic in an in Vitro Assay and in a Limited Short-Term
culture for the last 6 h. Cyclosporin (CsA) alone (■), CsA ϩ 250 nM TRAM-34
Acute in Vivo Toxicity Test. TRAM-34 (5 M) does not reduce cell
(ᮀ), CsA ϩ 500 nM TRAM-34 (Œ), CsA ϩ 1 M TRAM-34 (E).
viability of human T lymphocytes or several cell lines incubated
for 48 h with the compound (supplementary Table 5). Mice (n ϭ
5) injected intravenously with a single dose of TRAM-34 (0.5
Discussion
mg͞kg; 29 M) appeared clinically normal during the 7-day
Starting with clotrimazole, an azole antimycotic that blocks both
study. The body-weight data of the TRAM-34-treated group
the IKCa1 channel and mammalian cytochrome P450 enzymes at
(day 1:17.8 g; day 7: 27.0 g) were similar to control mice injected
nanomolar concentrations, we have developed compounds that
with the vehicle (day 1: 17.4 g; day 7: 23.4 g). Collectively, data
selectively target IKCa1. The pharmacophore for channel block
from these limited toxicity studies suggest that TRAM-34 is not
consists of a triphenyl moiety with an orthohalogen on one of the
PHARMACOLOGY
acutely toxic at Ϸ500–1,000 times the channel-blocking dose.
phenyl rings and substituted by a small unsubstituted polar
PNAS ͉ July 5, 2000 ͉ vol. 97 ͉ no. 14 ͉ 8155
-electron-rich heterocyle (pyrazole or tetrazole) or a nitrile
that is lined by residues from the cytoplasmic ends of S5 and S6
group (Fig. 2C). The molecular dimensions of this pharmaco-
and with dimensions to match the estimated size of the triphe-
phore are Ϸ9.5 Å by 9.5Å by 8.6 Å, giving a molecular volume
of 308 Å3. Smaller molecules that keep the perfect propeller
The most potent channel inhibitor, TRAM-34 (Kd ϭ 20 nM),
shape of the molecule retain potency (5a and 5b), whereas the
exhibits a 200- to 1,500-fold selectivity for IKCa1 over Kv, BKCa,
introduction of even small substituents such as a methyl group
SKCa, Na, CRAC, and chloride channels, and unlike clotrim-
(6h, 6i) on the heterocycle lower potency by increasing size.
azole does not inhibit the major mammalian cytochrome P450
Replacing the heterocycle with nonaromatic substituents (e.g.,
enzyme, CYP3A4. TRAM-34 also does not exhibit toxicity in an
2a, 3a, 3e, 3h) greatly reduces activity, the only exception being in vitro assay or cause obvious deleterious changes in a limited
the nitrile group (5a, 5b) that has a -electron density similar to
short-term acute toxicity study in rodents. [3H]thymidine incor-
imidazole (Figs. 1 and 2B). Other substitutions that alter the
poration assays using TRAM-34 as a selective inhibitor of IKCa1
-electron density in the heterocycle (6j) and͞or distort the
demonstrate that the channel plays an important role in the
molecular shape (6k) also reduce potency. Affinity of these
reactivation process of human lymphocytes. IKCa1 blockers
compounds for the channel does not correlate with their lipophi-
might therefore have use for the treatment of diverse autoim-
licity (supplementary Fig. 6, www.pnas.org). From these struc-
mune disorders in which reactivation of T lymphocytes contrib-
ture-activity relationships, we postulate that triphenylmethanes
utes to the pathogenesis of the disease. Because TRAM-34 and
bind to a size-restricted pocket in the IKCa1 channel, possibly via
cyclosporin A suppress T-cell mitogenesis more potently than
– electron interactions involving the three phenyl rings and
either compound alone, IKCa1 blockers may be useful for
the pyrazole, tetrazole, or imidazole moiety. Another possibility
combination therapy to reduce cyclosporin A toxicity. These
is that the benzphenone phenyl groups do not participate in
encouraging results suggest that TRAM-34 should be further
binding but instead serve as a scaffold, holding the -bonded
evaluated for possible therapeutic applications. TRAM-34 also
nitrogen, quaternary carbon, and ortho-halogen in place (Fig. 2C).
has immediate value as a pharmacological tool to define the role
Clotrimazole and the related triarylmethanes, although ap-
of IKCa1 channels in human tissues.
plied externally in our studies, should readily cross the cell
membrane because of their lipophilicity (clotrimazole: logP: 3.5;
We thank Dr. Luette Forrest, Ms. Chialing Wu, Elke Stoll, and Susan
TRAM-34 logP: 4.0) and may interact with a site on the inner
Ha¨uer for their excellent technical assistance. We are also indebted to
surface of the channel, possibly accounting for the slow onset of
Dr. Dieter Heber for advice on chemical nomenclature, to Dr. Ulrich
block. Consistent with this idea, an earlier study with a mem-
Girreser for NMR and mass spectrometry, to Dr. Hubert Kerschbaum
brane-impermeant quaternary derivative of clotrimazole re-
for the CRAC channel experiments, and to Dr. Heiko Rauer for
electrophysiological analysis of four initial compounds. This research was
vealed an internal binding site on the IKCa1 channel (37). A
funded by National Institutes of Health Grants MH59222 (K.G.C.) and
molecular model of the IKCa1 inner vestibule (38) based on the
NS 14069 (M.D.C.) and by a fellowship grant (WU 320͞1–1) from the
KcsA crystal structure (39) contains a putative binding pocket
Deutsche Forschungsgemeinschaft (H.W.).
1. Rodrigues, A. D., Gibson, G. G., Ioannides, C. & Parke, D. V. (1987) Biochem.
18. Goodman, A. G., Rali, T. W., Nies, A. S. & Taylor, P. (1990) Goodman andPharmacol. 36, 4277–4281. Gilman’s The Pharmacological Basis of Therapeutics 1169–1677.
2. Ayub, M. & Levell, M. J. (1990) Biochem. Pharmacol. 40, 1569–1775.
19. Cahalan, M. D. & Chandy, K. G. (1997) Curr. Opin. Biotechnol. 8, 749–756.
3. Maurice, M., Pichard, L., Daujat, M., Fabre, I., Joyeux, H., Domergue, J. &
20. Grissmer, S., Nguyen, A. N. & Cahalan, M. D. (1993) J. Gen. Physiol. 102,
Maurel, P. (1992) FASEB J. 6, 752–758.
4. Fowler, S. M., Riley, R. J., Pritchard, M. P., Sutcliffe, M. J., Friedberg, T. &
21. Hess, S. D., Oortgiesen, M. & Cahalan, M. D. (1993) J. Immunol. 150,
Wolf, R. C. (2000) Biochemistry 39, 4406–4414.
5. Ishii, T. M., Silvia, C., Hirschberg, B., Bond, C. T., Adelman, J. P. & Maylie,
22. Verheugen, J. A., Le Deist, F., Devignot, V. & Korn, H. (1997) Cell Calcium
J. (1997) Proc. Natl. Acad. Sci. USA 94, 11651–11656. 21, 1–17.
6. Joiner, W. J., Wang, L. Y., Tang, M. D. & Kaczmarek, L. K. (1997) Proc. Natl.
23. Bu¨chel, K. H., Draber, W., Regel, E. & Plempel, M. (1972) Arneim.-Forsch. 22, Acad. Sci. USA 94, 11013–11018.
7. Logsdon, N. J., Kang, J., Togo, J. A., Christian, E. P. & Aiyar, J. (1997) J. Biol.
24. Bartroli, J., Alguero, M., Boncompte, E. & Forn, J. (1992) Arzneim.-Forsch. 42, Chem. 272, 32723–32726.
8. Alvarez, J., Montero, M. & Garcia-Sancho, J. (1992) J. Biol. Chem. 267,
25. Ng, K.-K. D. & Hart, H. (1995) Tetrahedron 51, 7883–7906.
26. Casadio, S., Donetti, A. & Coppi, G. (1973) J. Pharm. Sci. 62, 773–778.
9. Brugnara, C., Gee, B., Armsby, C. C., Kurth, S., Sakamoto, M., Rifai, N., Alper,
27. Loch, G. & Rieger, V. (1953) Chem. Ber. 86, 74–76.
S. L. & Platt, O. S. (1996) J. Clin. Invest. 97, 1227–1234.
28. Grissmer, S., Nguyen, A. N., Aiyar, J., Hanson, D. C., Mather, R. J., Gutman,
10. Vandorpe, D. H., Shmukler, B. E., Jiang, L., Lim, B., Maylie, J., Adelman, J. P.,
G. A., Karmilowicz, M. J., Auperin, D. D. & Chandy, K. G. (1994) Mol.
de Franceschi, L., Cappellini, M. D., Brugnara, C. & Alper, S. L. (1998) J. Biol.Pharmacol. 45, 1227–1234. Chem. 273, 21542–21553.
29. Ja¨ger, H., Adelman, J. P. & Grissmer, S. (2000) FEBS Lett. 469, 196–202.
11. Fanger, C. M., Ghanshani, S., Logsdon, N. J., Rauer, H., Kalman, K., Zhou,
30. Wilson, A. J., Tinker, A. & Clapp, L. H. (1999) The Physiologist 42, A7.
J., Beckingham, K., Chandy, K. G., Cahalan, M. D. & Aiyar, J. (1999) J. Biol.
31. Kerschbaum, H. & Cahalan, M. D. (1999) Science 283, 836–839. Chem. 274, 5746–5754.
32. Ross, P. E., Garber, S. S. & Cahalan, M. D. (1994) Biophys. J. 66, 169–178.
12. Khanna, R., Chang, M. C., Joiner, W. J., Kaczmarek, L. K. & Schlichter, L. C.
33. Henderson, G. L., Harkey, M. R., Gershwin, M. E., Hackman, R. M., Stern, J. S.
(1999) J. Biol. Chem. 274, 14838–14849.
& Stressser, D. M. (1999) Life Sci. 65, PL209–214.
13. Jensen, B. S., Odum, N., Jorgensen, N. K., Christophersen, P. & Olesen, S. P.
34. Gibbs, M. A., Kunze, K. L., Howold, W. N. & Thummel, K. E. (1999) Drug
(1999) Proc. Natl. Acad. Sci. USA 96, 10917–10921. Metab. Dispos. 27, 596–599.
14. Rufo, P. A., Merlin, D., Riegler, M., Ferguson-Maltzman, M. H., Dickinson,
35. Song, H. & Shin, H.-S. (1998) Acta Crystallogr. C 54, 1675–1677.
B. L., Brugnara, C., Alper, S. L. & Lencer, W. I. (1997) J. Clin. Invest. 100,
36. Kalman, K., Pennington, M. W., Lanigan, M. D., Nguyen, A., Rauer, H.,
Mahnir, V., Paschetto, K., Kem, W. R., Grissmer, S., Gutman, G. A., et al.
15. Wojtulewski, J. A., Gow, P. J., Walter, J., Grahame, R., Gibson, T., Panayi, G. S.
(1998) J. Biol. Chem. 273, 32697–32707.
& Mason, J. (1980) Ann. Rheum. Dis. 39, 469–472.
37. Dunn, P. M. (1998) J. Membr. Biol. 165, 133–143.
16. Sawyer, P. R., Brogden, R. N., Pinder, R. M., Speight, T. M. & Avery, G. S.
38. Rauer, H., Pennington, M., Cahalan, M. & Chandy, K. G. (1999) J. Biol. Chem.
(1975) Drugs 9, 424–447. 274, 21885–21892.
17. Slama, J. T., Hancock, J. L., Rho, T., Sambucci, L. & Bachmann, K. A. (1998)
39. Doyle, D. A., Morais Cabral, J., Pfuetzner, R. A., Kuo, A., Gulbis, J. M., Cohen,
Biochem. Pharmacol. 55, 1881–1892.
S. L., Chait, B. T. & MacKinnon, R. (1998) Science 280, 69–77. 8156 ͉ www.pnas.org
BRETT L. SCHMIDLI INDUSTRIAL EXPERIENCE Beckman Coulter, Inc. Vice President, Discovery & Lab Automation Supply Chain Management 8/09 to Present Vice President, Immunoassay Manufacturing Operations 6/07 to 8/09 I lead the High Sensitivity Testing (HST) Group supply chains for our Immunoassay, Manual Immunoassay, and Molecular Diagnostics businesses. This in
T. Boone Pickens Media Coverage 4.10.10-4.12.10 Total of 20 Placements Coverage Summary: US News & World Report has a special issue dedicated to energy in which Pickens is mentioned in two pieces. The first article discusses the progress that is being made to move towards clean energy sources, but says more still needs to be done. The article mentions the components of the P
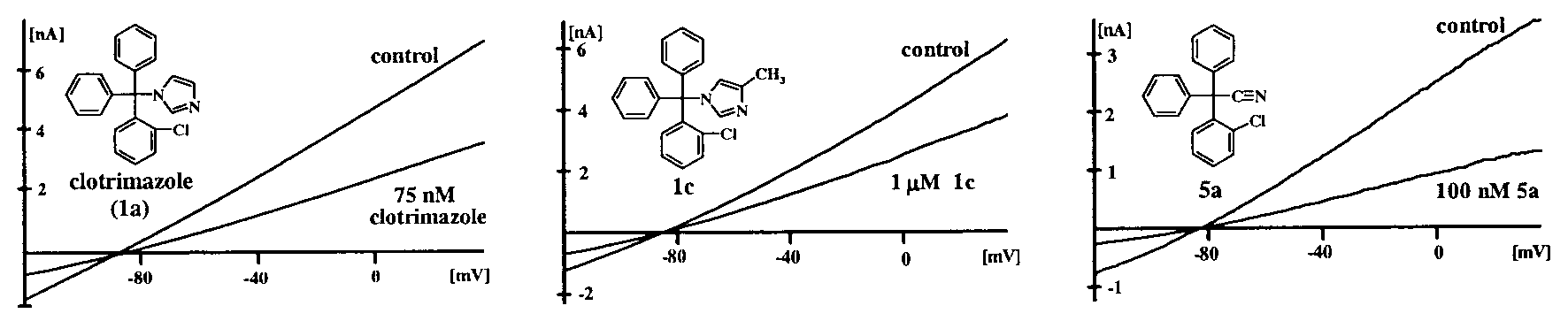 Inhibitory effects of compounds 1a (clotrimazole), 1c, and 5a on IKCa1 currents expressed in COS-7 cells. Voltage ramps were applied from Ϫ120 mV
Inhibitory effects of compounds 1a (clotrimazole), 1c, and 5a on IKCa1 currents expressed in COS-7 cells. Voltage ramps were applied from Ϫ120 mV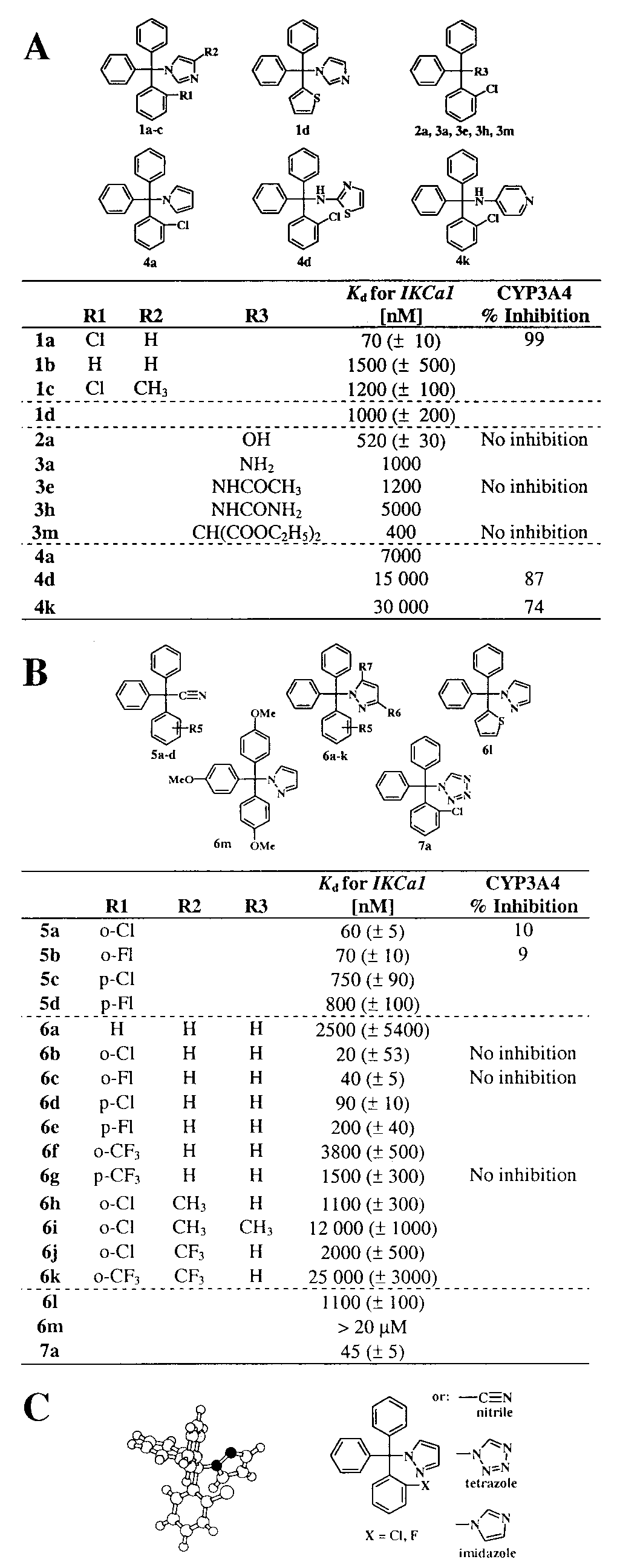 blocking potencies of 30 exemplary compounds that highlight
our design strategy are described in Fig. 2, and their physical data
are listed in supplementary Table 3 (www.pnas.org). The struc-
tures and channel affinities of the remaining 53 compounds are
provided in supplementary Table 3 and their physical data in
supplementary Table 4. The hydrolytic stability of TRAM-34 is
To test whether the imidazole ring is necessary for channel
blocking activity, we generated several analogs where this moiety
is replaced by a hydroxyl- (2a-p), an amino- (3a-d), an acet-
blocking potencies of 30 exemplary compounds that highlight
our design strategy are described in Fig. 2, and their physical data
are listed in supplementary Table 3 (www.pnas.org). The struc-
tures and channel affinities of the remaining 53 compounds are
provided in supplementary Table 3 and their physical data in
supplementary Table 4. The hydrolytic stability of TRAM-34 is
To test whether the imidazole ring is necessary for channel
blocking activity, we generated several analogs where this moiety
is replaced by a hydroxyl- (2a-p), an amino- (3a-d), an acet-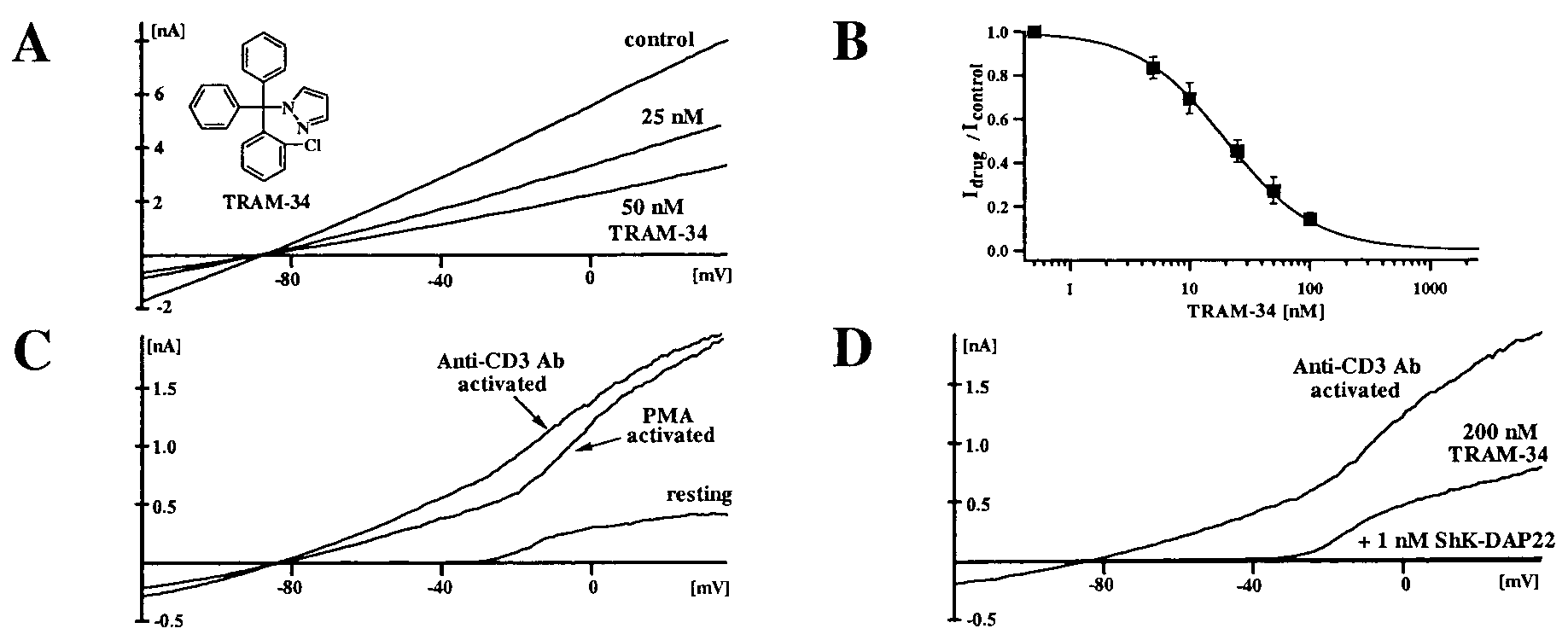 Table 1. Selectivity
Table 1. Selectivity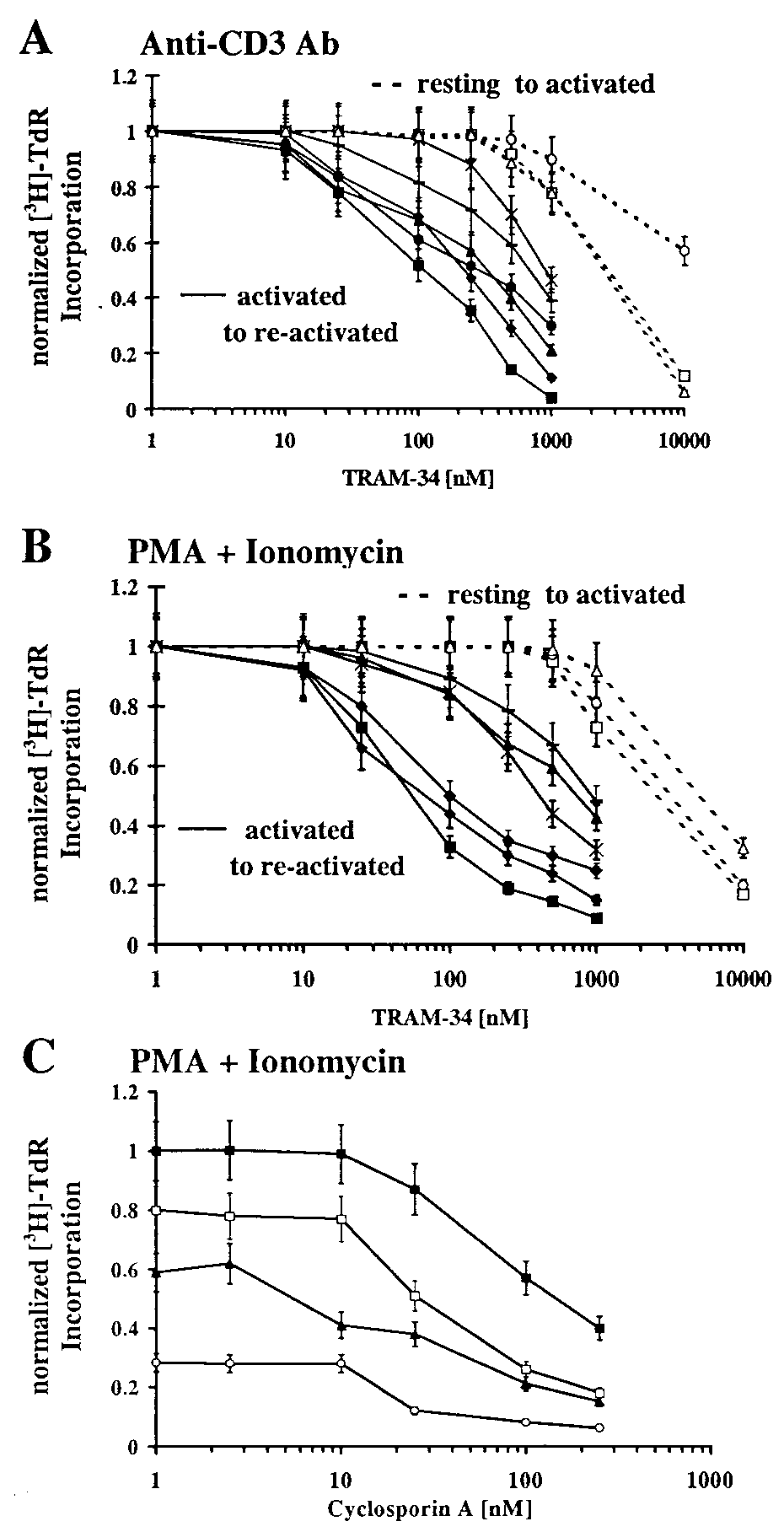 gen- and mitogen-induced proliferation of resting human lym-
phocytes. Since this concentration is Ϸ100 times the channel-
blocking dose, suppression is probably due to a nonspecific
mechanism. Studies done at the same time by Khanna et al. (12)
showed that 250 nM clotrimazole (a concentration closer to the
channel-blocking dose) suppresses the activation of phytohe-
magglutinin-preactivated T cells more effectively than the acti-
vation of quiescent cells. However, because clotrimazole blocks
both IKCa1 and cytochrome P450 enzymes, the mechanism
underlying this suppression remains unclear.
gen- and mitogen-induced proliferation of resting human lym-
phocytes. Since this concentration is Ϸ100 times the channel-
blocking dose, suppression is probably due to a nonspecific
mechanism. Studies done at the same time by Khanna et al. (12)
showed that 250 nM clotrimazole (a concentration closer to the
channel-blocking dose) suppresses the activation of phytohe-
magglutinin-preactivated T cells more effectively than the acti-
vation of quiescent cells. However, because clotrimazole blocks
both IKCa1 and cytochrome P450 enzymes, the mechanism
underlying this suppression remains unclear.