Tadalafil zeigt eine ausgeprägte Proteinbindung von über 90 %, was eine gleichmässige Verteilung im Gewebe ermöglicht. Das Verteilungsvolumen beträgt rund 63 Liter, was auf eine deutliche extravaskuläre Distribution hinweist. Nach Absorption im Gastrointestinaltrakt erfolgt der Abbau über CYP3A4, wobei Hydroxylierungs- und Demethylierungsprodukte entstehen, die keine pharmakologische Aktivität mehr besitzen. Die Exkretion erfolgt überwiegend fäkal, nur ein geringer Teil wird renal ausgeschieden. Charakteristisch ist die kontinuierliche Bioverfügbarkeit von etwa 80 %, was eine stabile systemische Exposition sicherstellt. Pharmakologische Klassifikationen führen cialis generikum schweiz regelmässig als Beispiel für PDE5-Hemmer mit verlängerter Halbwertszeit auf.
Im4676.pdf
VIOXX®(rofecoxib tablets and oral suspension) DESCRIPTION
VIOXX* (rofecoxib) is described chemically as 4-[4-(methylsulfonyl)phenyl]-3-phenyl-
2(5H)-furanone. It has the following chemical structure:
Rofecoxib is a white to off-white to light yellow powder. It is sparingly soluble in acetone,
slightly soluble in methanol and isopropyl acetate, very slightly soluble in ethanol,
practically insoluble in octanol, and insoluble in water. The empirical formula for
rofecoxib is C17H14O4S, and the molecular weight is 314.36.
Each tablet of VIOXX for oral administration contains either 12.5 or 25 mg of rofecoxib
and the following inactive ingredients: croscarmellose sodium, hydroxypropyl cellulose,
lactose, magnesium stearate, microcrystalline cellulose, and yellow ferric oxide.
Each 5 mL of the oral suspension contains either 12.5 or 25 mg of rofecoxib and the
following inactive ingredients: citric acid (monohydrate), sodium citrate (dihydrate),
sorbitol solution, strawberry flavor, xanthan gum, and purified water. Added as
preservatives are sodium methylparaben 0.13% and sodium propylparaben 0.02%. CLINICAL PHARMACOLOGY Mechanism of Action
VIOXX is a nonsteroidal anti-inflammatory drug that exhibits anti-inflammatory,
analgesic, and antipyretic activities in animal models. The mechanism of action of VIOXX
is believed to be due to inhibition of prostaglandin synthesis, via inhibition of
cyclooxygenase-2 (COX-2). At therapeutic concentrations in humans, VIOXX does not
inhibit the cyclooxygenase-1 (COX-1) isoenzyme. Pharmacokinetics
* Registered trademark of MERCK & CO., Inc., Whitehouse Station, New Jersey, USA COPYRIGHT MERCK & CO., Inc., 1998 All rights reserved. Absorption
The mean oral bioavailability of VIOXX at therapeutically recommended doses of 12.5, 25
and 50 mg is approximately 93%. The area under the curve (AUC) and peak plasma level
(Cmax) following a single 25 mg dose were 3286( ±843) ng*hr/mL and 207 (±111) ng/mL,
respectively. Both Cmax and AUC are roughly dose proportional across the clinical dose
range. At doses greater than 50 mg, there is a less than proportional increase in Cmax and
AUC, which is thought to be due to the low solubility of the drug in aqueous media. The
plasma concentration-time profile exhibited multiple peaks. The median time to maximal
concentration (Tmax), as assessed in nine pharmacokinetic studies, is 2 to 3 hours.
Individual Tmax values in these studies ranged between 2 to 9 hours. This may not reflect
rate of absorption as Tmax may occur as a secondary peak in some individuals. With
multiple dosing, steady state conditions are reached by Day 4. The AUC0-24hr and Cmax at
steady-state after multiple doses of 25 mg rofecoxib was 4018 (±1140) ng∗hr/mL and 321 (±104)
ng/mL, respectively. The accumulation factor based on geometric means was 1.67.
VIOXX Tablets and VIOXX Oral Suspension are bioequivalent. Food and Antacid Effects
Food had no significant effect on either the peak plasma concentration (Cmax) or extent of absorption
(AUC) of rofecoxib when VIOXX tablets were taken with a high fat meal. The time to peak plasma
concentration (Tmax), however, was delayed by 1 to 2 hours. The food effect on the suspension
formulation has not been studied. VIOXX tablets can be administered without regard to timing of meals.
There was a 13% and 8% decrease in AUC when VIOXX was administered with calcium
carbonate antacid and magnesium/aluminum antacid to elderly subjects, respectively.
There was an approximate 20% decrease in Cmax of rofecoxib with either antacid. Distribution
Rofecoxib is approximately 87% bound to human plasma protein over the range of
concentrations of 0.05 to 25 µg/mL. The apparent volume of distribution at steady state
(Vdss) is approximately 91 L following a 12.5 mg dose and 86 L following a 25 mg dose.
Rofecoxib has been shown to cross the placenta in rats and rabbits, and the blood-brain
Metabolism
Metabolism of rofecoxib is primarily mediated through reduction by cytosolic enzymes. The principal
metabolic products are the cis-dihydro and trans-dihydro derivatives of rofecoxib, which account for
nearly 56% of recovered radioactivity in the urine. An additional 8.8% of the dose was recovered as the
glucuronide of the hydroxy derivative, a product of oxidative metabolism. The biotransformation of
rofecoxib and this metabolite is reversible in humans to a limited extent (<5%). These metabolites are
inactive as COX-1 or COX-2 inhibitors.
Cytochrome P450 plays a minor role in metabolism of rofecoxib. Inhibition of CYP3A activity by
administration of ketoconazole 400 mg daily does not affect rofecoxib disposition. However, induction of
general hepatic metabolic activity by administration of the non-specific inducer rifampin 600 mg daily
produces a 50% decrease in rofecoxib plasma concentrations. (Also see Drug Interactions). Excretion
Rofecoxib is eliminated predominantly by hepatic metabolism with little (<1%) unchanged
drug recovered in the urine. Following a single radiolabeled dose of 125 mg,
approximately 72% of the dose was excreted into the urine as metabolites, and 14% in the
The plasma clearance after 12.5 and 25 mg doses was approximately 141 and 120
mL/min, respectively. Higher plasma clearance was observed at doses below the
therapeutic range, suggesting the presence of a saturable route of metabolism (i.e, non-
linear elimination). The effective half-life (based on steady state levels) was approximately
Special Populations Gender
The pharmacokinetics of rofecoxib are comparable in men and women. Geriatric
After a single dose of 25 mg VIOXX in elderly subjects (over 65 years old) a 34%
increase in AUC was observed as compared to the young subjects. Dosage adjustment in
the elderly is not necessary; however, therapy with VIOXX should be initiated at the
Pediatric
VIOXX has not been investigated in patients below 18 years of age. Race
Meta-analysis of pharmacokinetic studies has suggested a slightly (10-15%) higher AUC
of rofecoxib in Blacks and Hispanics as compared to Caucasians. No dosage adjustment
Hepatic Insufficiency
A pharmacokinetic study in mild (Child-Pugh score <6) hepatic insufficiency patients
indicated that rofecoxib AUC was similar between these patients and healthy subjects.
Limited data in patients with moderate (Child-Pugh score 7-9) hepatic insufficiency
suggest a trend towards higher AUC (about 69%) of rofecoxib in these patients, but more
data are needed to evaluate pharmacokinetics in these patients. Patients with severe
hepatic insufficiency have not been studied. Renal Insufficiency
In a study (N=6) of patients with end stage renal disease undergoing dialysis, peak
rofecoxib plasma levels and AUC declined 18% and 9%, respectively, when dialysis
occurred four hours after dosing. When dialysis occurred 48 hours after dosing, the
elimination profile of rofecoxib was unchanged. While renal insufficiency does not
influence the pharmacokinetics of rofecoxib, use of VIOXX in advanced renal disease is
not recommended at present because no safety information is available regarding the use
Drug Interactions
Also see PRECAUTIONS – Drug Interactions. General: In human studies the potential for rofecoxib to inhibit or induce CYP 3A4
activity was investigated in studies using the intravenous erythromycin breath test and the
oral midazolam test. No significant difference in erythromycin demethylation was observed
with rofecoxib (75 mg daily) compared to placebo, indicating no induction of hepatic CYP
3A4. A 30% reduction of the AUC of midazolam was observed with rofecoxib (25 mg
daily). This reduction is most likely due to increased first pass metabolism through
induction of intestinal CYP 3A4 by rofecoxib. In vitro studies in rat hepatocytes also
suggest thatrofecoxib might be a mild inducer for CYP 3A4.
Drug interaction studies with rofecoxib have identified potentially significant interactions
with rifampin, methotrexate and warfarin. Patients receiving these agents with VIOXX
should be appropriately monitored. Drug interaction studies do not support the potential
for clinically important interactions between antacids or cimetidine with rofecoxib. Similar
to experience with other nonsteroidal antiinflammatory drugs (NSAIDs), studies with
rofecoxib suggest the potential for interaction with ACE inhibitors. The effects of
rofecoxib on the pharmacokinetics and/or pharmacodynamics of ketoconazole,
prednisone/prednisolone, oral contraceptives, and digoxin have been studied in vivo and
clinically important interactions have not been found . CLINICAL STUDIES Osteoarthritis (OA)
VIOXX has demonstrated significant reduction in joint pain compared to placebo.
VIOXX was evaluated for the treatment of the signs and symptoms of OA of the knee and
hip in placebo- and active-controlled clinical trials of 6 to 86 weeks duration that enrolled
approximately 3900 patients. In patients with OA, treatment with VIOXX 12.5 mg and
25 mg once daily resulted in improvement in patient and physician global assessments and
in the WOMAC (Western Ontario and McMaster Universities) osteoarthritis
questionnaire, including pain, stiffness, and functional measures of OA. In six studies of
pain accompanying OA flare, VIOXX provided a significant reduction in pain at the first
determination (after one week in one study, after two weeks in the remaining five studies);
this continued for the duration of the studies. In all OA clinical studies, once daily
treatment in the morning with VIOXX 12.5 and 25 mg was associated with a significant
reduction in joint stiffness upon first awakening in the morning. At doses of 12.5 and 25
mg, the effectiveness of VIOXX was shown to be comparable to ibuprofen 800 mg TID
and diclofenac 50 mg TID for treatment of the signs and symptoms of OA. The ibuprofen
studies were 6 week studies; the diclofenac studies were 12 month studies in which
patients could receive additional arthritis medication during the last 6 months. Analgesia, including Dysmenorrhea
In acute analgesic models of post-operative dental pain, post-orthopedic surgical pain, and
primary dysmenorrhea, VIOXX relieved pain that was rated by patients as moderate to
severe. The analgesic effect (including onset of action) of a single 50-mg dose of VIOXX
was generally similar to 550 mg of naproxen sodium or 400 mg of ibuprofen. In single-
dose post-operative dental pain studies, the onset of analgesia with a single 50 mg dose of
VIOXX occurred within 45 minutes. In a multiple-dose study of post-orthopedic surgical
pain in which patients received VIOXX or placebo for up to 5 days, 50 mg of VIOXX
once daily was effective in reducing pain. In this study, patients on VIOXX consumed a
significantly smaller amount of additional analgesic medication than patients treated with
placebo (1.5 versus 2.5 doses per day of additional analgesic medication for VIOXX and
Special Studies Upper Endoscopy in Patients with Osteoarthritis:
Two identical (U.S. and Multinational) endoscopy studies in a total of 1516 patients were
conducted to compare the percentage of patients who developed endoscopically
detectable gastroduodenal ulcers with VIOXX 25 mg daily or 50 mg daily, ibuprofen 2400
mg daily, or placebo. Entry criteria for these studies permitted enrollment of patients with
active Heliobacter pylori infection, baseline gastroduodenal erosions, prior history of an
upper gastrointestinal perforation, ulcer, or bleed (PUB), and/or age > 65 years. However,
patients receiving aspirin (including low-dose aspirin for cardiovascular prophylaxis) were
not enrolled in these studies. Patients who were 50 years of age and older with
osteoarthritis and who had no ulcers at baseline were evaluated by endoscopy after weeks
6, 12, and 24 of treatment. The placebo-treatment group was discontinued at week 16 by
Treatment with VIOXX 25 mg daily or 50 mg daily was associated with a significantly
lower percentage of patients with endoscopic gastroduodenal ulcers than treatment with
ibuprofen 2400 mg daily. However, the studies cannot rule out at least some increase in
the rate of endoscopic gastroduodenal ulcers when comparing VIOXX to placebo. See
Figures 1 and 2 and accompanying Tables for the results of these studies. COMPARISON TO IBUPROFEN Life-Table Cumulative Incidence Rate of Gastroduodenal Ulcers ≥ 3mm** (Intention-to-Treat)
** Results of analyses using a ≥ 5mm gastroduodenal ulcer endpoint were consistent. *** The primary endpoint was the cumulative incidence of gastroduodenal ulcer at 12 weeks. Endoscopic Gastroduodenal Ulcers at 12 weeks U.S. Study Number of Patients Cumulative Treatment with Ulcer/Total Incidence Number of Patients Ratio of Rates COMPARISON TO IBUPROFEN Life-Table Cumulative Incidence Rate of Gastroduodenal Ulcers ≥ 3mm** (Intention-to-Treat)
Results of analyses using a ≥ 5mm gastroduodenal ulcer endpoint were consistent.
The primary endpoint was the cumulative incidence of gastroduodenal ulcer at 12 weeks. Endoscopic Gastroduodenal Ulcers at 12 weeks Multinational Study Number of Patients Cumulative Treatment with Ulcer/Total Incidence Number of Patients Ratio of Rates
The correlation between findings of endoscopic studies, and the relative incidence of
clinically serious upper GI events that may be observed with different products, has not
been fully established. Serious clinically significant upper GI bleeding has been observed
in patients receiving VIOXX in controlled trials, albeit infrequently (see WARNINGS –
Gastrointestinal [GI] Effects). Prospective, long-term studies required to compare the
incidence of serious, clinically significant upper GI adverse events in patients taking
VIOXX versus comparator NSAID products have not been performed. Assessment of Fecal Occult Blood Loss in Healthy Subjects:
Occult fecal blood loss associated with VIOXX 25 mg daily, VIOXX 50 mg daily,
ibuprofen 2400 mg per day, and placebo was evaluated in a study utilizing 51Cr-tagged red
blood cells in 67 healthy males. After 4 weeks of treatment with VIOXX 25 mg daily or
VIOXX 50 mg daily, the increase in the amount of fecal blood loss was not statistically
significant compared with placebo-treated subjects. In contrast, ibuprofen 2400 mg per
day produced a statistically significant increase in fecal blood loss as compared with
placebo-treated subjects and VIOXX-treated subjects. The clinical relevance of this
Platelets:
Multiple doses of VIOXX 12.5, 25, and up to 375 mg administered daily up to 12 days
had no effect on bleeding time relative to placebo. Similarly, bleeding time was not altered
in a single dose study with 500 or 1000 mg of VIOXX. There was no inhibition of ex vivo
arachidonic acid- or collagen-induced platelet aggregation with 12.5, 25, and 50 mg of
INDICATIONS AND USAGE
For relief of the signs and symptoms of osteoarthritis.
For the management of acute pain in adults (see CLINICAL STUDIES).
For the treatment of primary dysmenorrhea. CONTRAINDICATIONS
VIOXX is contraindicated in patients with known hypersensitivity to rofecoxib or any
VIOXX should not be given to patients who have experienced asthma, urticaria, or
allergic-type reactions after taking aspirin or other NSAIDs. Severe, rarely fatal,
anaphylactic-like reactions to NSAIDs have been reported in such patients (see
WARNINGS - Anaphylactoid Reactions, and PRECAUTIONS - Preexisting Asthma). WARNINGS Gastrointestinal (GI) Effects - Risk of GI Ulceration, Bleeding, and Perforation:
Serious gastrointestinal toxicity such as bleeding, ulceration, and perforation of the
stomach, small intestine or large intestine, can occur at any time, with or without warning
symptoms, in patients treated with nonsteroidal anti-inflammatory drugs (NSAIDs).
Minor upper gastrointestinal problems, such as dyspepsia, are common and may also
occur at any time during NSAID therapy. Therefore, physicians and patients should
remain alert for ulceration and bleeding, even in the absence of previous GI tract
symptoms. Patients should be informed about the signs and/or symptoms of serious GI
toxicity and the steps to take if they occur. The utility of periodic laboratory monitoring
has not been demonstrated, nor has it been adequately assessed. Only one in five patients
who develop a serious upper GI adverse event on NSAID therapy is symptomatic. It has
been demonstrated that upper GI ulcers, gross bleeding or perforation, caused by
NSAIDs, appear to occur in approximately 1% of patients treated for 3-6 months, and in
about 2-4% of patients treated for one year. These trends continue thus, increasing the
likelihood of developing a serious GI event at some time during the course of therapy.
However, even short-term therapy is not without risk.
It is unclear, at the present time, how the above rates apply to VIOXX (see CLINICAL
STUDIES, Special Studies, Upper Endoscopy in Patients with Osteoarthritis). Among
3357 patients who received VIOXX in controlled clinical trials of 6 weeks to one year
duration (most were enrolled in six month or longer studies) at a daily dose of 12.5 mg to
50 mg, a total of 4 patients experienced a serious upper GI event, using protocol derived
criteria. Two patients experienced an upper GI bleed within three months (at day 62 and
87 respectively) (0.06%). One additional patient experienced an obstruction within six
months (Day 130) and the remaining patient developed an upper GI bleed within 12
months (Day 322) (0.12%). Approximately 23% of these 3357 patients were in studies
that required them to be free of ulcers at study entry. It is unclear if this study population
is representative of the general population. Prospective, long-term studies required to
compare the incidence of serious, clinically significant upper GI adverse events in patients
taking VIOXX vs comparator NSAID products have not been performed.
NSAIDs should be prescribed with extreme caution in patients with a prior history of ulcer
disease or gastrointestinal bleeding. Most spontaneous reports of fatal GI events are in
elderly or debilitated patients and therefore special care should be taken in treating this
population. To minimize the potential risk for an adverse GI event, the lowest effective dose should be used for the shortest possible duration. For high risk patients,
alternate therapies that do not involve NSAIDs should be considered.
Studies have shown that patients with a prior history of peptic ulcer disease and/orgastrointestinal bleeding and who use NSAIDs, have a greater than 10-fold higher risk for
developing a GI bleed than patients with neither of these risk factors. In addition to a past
history of ulcer disease, pharmacoepidemiological studies have identified several other co-
therapies or co-morbid conditions that may increase the risk for GI bleeding such as:
treatment with oral corticosteroids, treatment with anticoagulants, longer duration of
NSAID therapy, smoking, alcoholism, older age, and poor general health status. Anaphylactoid Reactions
Anaphylactoid reactions were not reported in patients receiving VIOXX in clinical trials.
However, as with NSAIDs in general, anaphylactoid reactions may occur in patients
without known prior exposure to VIOXX. VIOXX should not be given to patients with
the aspirin triad. This symptom complex typically occurs in asthmatic patients who
experience rhinitis with or without nasal polyps, or who exhibit severe, potentially fatal
bronchospasm after taking aspirin or other NSAIDs (see CONTRAINDICATIONS and
PRECAUTIONS - Preexisting Asthma). Emergency help should be sought in cases where
Advanced Renal Disease
No safety information is available regarding the use of VIOXX in patients with advanced
kidney disease. Therefore, treatment with VIOXX is not recommended in these patients.
If VIOXX therapy must be initiated, close monitoring of the patient's kidney function is
advisable (see PRECAUTIONS - Renal Effects). Pregnancy
In late pregnancy VIOXX should be avoided because it may cause premature closure of
PRECAUTIONS
VIOXX cannot be expected to substitute for corticosteroids or to treat corticosteroid
insufficiency. Abrupt discontinuation of corticosteroids may lead to exacerbation of
corticosteroid-responsive illness. Patients on prolonged corticosteroid therapy should
have their therapy tapered slowly if a decision is made to discontinue corticosteroids.
The pharmacological activity of VIOXX in reducing inflammation, and possibly fever, may
diminish the utility of these diagnostic signs in detecting infectious complications of
presumed noninfectious, painful conditions. Hepatic Effects:
Borderline elevations of one or more liver tests may occur in up to 15% of patients taking
NSAIDs, and notable elevations of ALT or AST (approximately three or more times the
upper limit of normal) have been reported in approximately 1% of patients in clinical trials
with NSAIDs. These laboratory abnormalities may progress, may remain unchanged, or
may be transient with continuing therapy. Rare cases of severe hepatic reactions,
including jaundice and fatal fulminant hepatitis, liver necrosis and hepatic failure (some
with fatal outcome) have been reported with NSAIDs. In controlled clinical trials of
VIOXX, the incidence of borderline elevations of liver tests at doses of 12.5 and 25 mg
daily was comparable to the incidence observed with ibuprofen and lower than that
observed with diclofenac. In placebo-controlled trials, approximately 0.5% of patients
taking rofecoxib (12.5 or 25 mg QD) and 0.1% of patients taking placebo had notable
A patient with symptoms and/or signs suggesting liver dysfunction, or in whom an
abnormal liver test has occurred, should be monitored carefully for evidence of the
development of a more severe hepatic reaction while on therapy with VIOXX. Use of
VIOXX is not recommended in patients with moderate or severe hepatic insufficiency (see
Pharmacokinetics – Special Populations). If clinical signs and symptoms consistent with
liver disease develop, or if systemic manifestations occur (e.g., eosinophilia, rash, etc.),
Long-term administration of NSAIDs has resulted in renal papillary necrosis and other
renal injury. Renal toxicity has also been seen in patients in whom renal prostaglandins
have a compensatory role in the maintenance of renal perfusion. In these patients,
administration of a nonsteroidal anti-inflammatory drug may cause a dose-dependent
reduction in prostaglandin formation and, secondarily, in renal blood flow, which may
precipitate overt renal decompensation. Patients at greatest risk of this reaction are those
with impaired renal function, heart failure, liver dysfunction, those taking diuretics and
ACE inhibitors, and the elderly. Discontinuation of NSAID therapy is usually followed by
recovery to the pretreatment state. Clinical trials with VIOXX at daily doses of 12.5 and
25 mg have shown renal effects (e.g., hypertension, edema) similar to those observed with
comparator NSAIDs; these occur with an increased frequency with chronic use of VIOXX
at doses above the 12.5 to 25 mg range. (See ADVERSE REACTIONS.)
Caution should be used when initiating treatment with VIOXX in patients with
considerable dehydration. It is advisable to rehydrate patients first and then start therapy
with VIOXX. Caution is also recommended in patients with pre-existing kidney disease
(see WARNINGS-Advanced Renal Disease). Hematological Effects:
Anemia is sometimes seen in patients receiving VIOXX. In placebo-controlled trials,
there were no significant differences observed between VIOXX and placebo in clinical
reports of anemia. Patients on long-term treatment with VIOXX should have their
hemoglobin or hematocrit checked if they exhibit any signs or symptoms of anemia or
blood loss. VIOXX does not generally affect platelet counts, prothrombin time (PT), or
partial thromboplastin time (PTT), and does not inhibit platelet aggregation at indicated
dosages (See CLINICAL STUDIES-Special Studies-Platelets). Fluid Retention and Edema:
Fluid retention and edema have been observed in some patients taking VIOXX (see
ADVERSE REACTIONS). VIOXX should be used with caution, and should be
introduced at the lowest recommended dose, in patients with fluid retention,
Preexisting Asthma:
Patients with asthma may have aspirin-sensitive asthma. The use of aspirin in patients with
aspirin-sensitive asthma has been associated with severe bronchospasm which can be fatal.
Since cross reactivity, including bronchospasm, between aspirin and other nonsteroidal
anti-inflammatory drugs has been reported in such aspirin-sensitive patients, VIOXX
should not be administered to patients with this form of aspirin sensitivity and should be
used with caution in patients with preexisting asthma. Information for Patients
VIOXX can cause discomfort and, rarely, more serious side effects, such as
gastrointestinal bleeding, which may result in hospitalization and even fatal outcomes.
Although serious GI tract ulcerations and bleeding can occur without warning symptoms,
patients should be alert for the signs and symptoms of ulcerations and bleeding, and should
ask for medical advice when observing any indicative signs or symptoms. Patients should
be apprised of the importance of this follow-up (see WARNINGS, Gastrointestinal (GI)
Effects - Risk of GI Ulceration, Bleeding and Perforation).
Patients should promptly report signs or symptoms of gastrointestinal ulceration or
bleeding, skin rash, unexplained weight gain, or edema to their physicians.
Patients should be informed of the warning signs and symptoms of hepatotoxicity (e.g.,
nausea, fatigue, lethargy, pruritus, jaundice, right upper quadrant tenderness, and "flu-like"
symptoms). If these occur, patients should be instructed to stop therapy and seek
Patients should also be instructed to seek immediate emergency help in the case of an
anaphylactoid reaction (see WARNINGS).
In late pregnancy VIOXX should be avoided because it may cause premature closure of
Laboratory Tests
Because serious GI tract ulcerations and bleeding can occur without warning symptoms,
physicians should monitor for signs or symptoms of GI bleeding. Drug Interactions ACE-inhibitors: Reports suggest that NSAIDs may diminish the antihypertensive effect of
Angiotensin Converting Enzyme (ACE) inhibitors. In patients with mild to moderate
hypertension, administration of 25 mg daily of VIOXX with the ACE inhibitor benazepril,
10 to 40 mg for 4 weeks, was associated with an average increase in mean arterial
pressure of about 3 mm Hg compared to ACE inhibitor alone. This interaction should be
given consideration in patients taking VIOXX concomitantly with ACE-inhibitors. Aspirin: Concomitant administration of low-dose aspirin with VIOXX may result in an
increased rate of GI ulceration or other complications, compared to use of VIOXX alone.
At steady state, VIOXX 50 mg once daily had no effect on the anti-platelet activity of
low-dose (81 mg once daily) aspirin, as assessed by ex vivo platelet aggregation and serum
TXB2 generation in clotting blood. VIOXX is not a substitute for aspirin for
Cimetidine: Co-administration with high doses of cimetidine [800 mg twice daily]
increased the Cmax of rofecoxib by 21%, the AUC0-120hr by 23% and the t1/2 by 15%. These
small changes are not clinically significant and no dose adjustment is necessary. Digoxin: Rofecoxib 75 mg once daily for 11 days does not alter the plasma concentration
profile or renal elimination of digoxin after a single 0.5 mg oral dose. Furosemide: Clinical studies, as well as post marketing observations, have shown that
NSAIDs can reduce the natriuretic effect of furosemide and thiazides in some patients.
This response has been attributed to inhibition of renal prostaglandin synthesis. Ketoconazole: Ketoconazole 400 mg daily did not have any clinically important effect on Lithium: NSAIDs have produced an elevation of plasma lithium levels and a reduction in
renal lithium clearance. Thus, when VIOXX and lithium are administered concurrently,
subjects should be observed carefully for signs of lithium toxicity. Methotrexate: VIOXX 75 mg administered once daily for 10 days increased plasma
concentrations by 23% as measured by AUC0-24 hr in patients receiving methotrexate 7.5 to
15 mg/week for rheumatoid arthritis. An equivalent magnitude of reduction in
methotrexate renal clearance was observed. At 24 hours postdose, a similar proportion of
patients treated with methotrexate alone (94%) and subsequently treated with
methotrexate co-administered with 75 mg of rofecoxib (88%) had methotrexate plasma
concentrations below the measurable limit (5 ng/mL). The effects of the recommended
doses for osteoarthritis (12.5 and 25 mg) of VIOXX on plasma methotrexate levels are
unknown. Standard monitoring of methotrexate-related toxicity should be continued if
VIOXX and methotrexate are administered concomitantly. Oral Contraceptives: Rofecoxib did not have any clinically important effect on the
pharmacokinetics of ethinyl estradiol and norethindrone. Prednisone/prednisolone: Rofecoxib did not have any clinically important effect on the
pharmacokinetics of prednisolone or prednisone. Rifampin: Co-administration of VIOXX with rifampin 600mg daily, a potent inducer of
hepatic metabolism, produced an approximate 50% decrease in rofecoxib plasma
concentrations. Therefore, a starting daily dose of 25 mg of VIOXX should be considered
for the treatment of osteoarthritis when VIOXX is co-administered with potent inducers
Warfarin: Prothrombin time (measured as INR) increased in both single and multiple dose
cross-over studies in healthy individuals receiving both warfarin and rofecoxib. In a 21 day
multiple dose study in healthy individuals stabilized on warfarin (2 to 8.5 mg daily),
administration of rofecoxib 25 mg QD was associated with mean increases in INR of
approximately 8% (range of INR on warfarin alone, 1.1 to 2.2; range of INR on warfarin
plus rofecoxib, 1.2 to 2.4). Somewhat greater mean increases in INR of approximately
11% (range of maximum INR on warfarin alone, 1.5 to 2.7; range of maximum INR on
warfarin plus rofecoxib, 1.6 to 4.4) were also seen in a single dose PK screening study
using a 30 mg dose of warfarin and 50 mg of rofecoxib. Standard monitoring of INR
values should be conducted when therapy with VIOXX is initiated or changed, particularly
in the first few days, in patients receiving warfarin or similar agents. Carcinogenesis, Mutagenesis, Impairment of Fertility
Rofecoxib was not carcinogenic in mice given oral doses up to 30 mg/kg (male) and 60
mg/kg (female) (approximately 5- and 2-fold the human exposure at 25 and 50 mg daily
based on AUC0-24) and in male and female rats given oral doses up to 8 mg/kg
(approximately 6- and 2-fold the human exposure at 25 and 50 mg daily based on AUC0-
Rofecoxib was not mutagenic in an Ames test or in a V-79 mammalian cell mutagenesis
assay, nor clastogenic in a chromosome aberration assay in Chinese hamster ovary (CHO)
cells, in an in vitro and an in vivo alkaline elution assay, or in an in vivo chromosomal
aberration test in mouse bone marrow.
Rofecoxib did not impair male fertility in rats at oral doses up to 100 mg/kg
(approximately 20- and 7-fold human exposure at 25 and 50 mg daily based on the AUC0-
24) and rofecoxib had no effect on fertility in female rats at doses up to 30 mg/kg
(approximately 19- and 7-fold human exposure at 25 and 50 mg daily based on AUC0-24). Pregnancy: Teratogenic effects: Pregnancy Category C. Rofecoxib was not teratogenic in rats at
doses up to 50 mg/kg/day (approximately 28- and 10-fold human exposure at 25 and 50
mg daily based on AUC0-24). There was a slight, non-statistically significant increase in the
overall incidence of vertebral malformations only in the rabbit at doses of 50 mg/kg/day
(approximately 1- or <1-fold human exposure at 25 and 50 mg daily based on AUC0-24).
There are no studies in pregnant women. VIOXX should be used during pregnancy only if
the potential benefit justifies the potential risk to the fetus. Nonteratogenic effects: Rofecoxib produced peri-implantation and post-implantation
losses and reduced embryo/fetal survival in rats and rabbits at oral doses $10 and $75
mg/kg/day, respectively (approximately 9- and 3-fold [rats] and 2- and <1-fold [rabbits]
human exposure based on the AUC0-24 at 25 and 50 mg daily). These changes are
expected with inhibition of prostaglandin synthesis and are not the result of permanent
alteration of female reproductive function. There was an increase in the incidence of
postnatal pup mortality in rats at ≥5 mg/kg/day (approximately 5- and 2-fold human
exposure at 25 and 50 mg daily based on AUC0-24). ). In studies in pregnant rats
administered single doses of rofecoxib, there was a treatment-related decrease in the
diameter of the ductus arteriosus at all doses used (3-300 mg/kg: 3 mg/kg is
approximately 2- and <1-fold human exposure at 25 or 50 mg daily based on AUC0-24). As
with other drugs known to inhibit prostaglandin synthesis, use of VIOXX during the third
trimester of pregnancy should be avoided. Labor and delivery: Rofecoxib produced no evidence of significantly delayed labor or
parturition in females at doses <15 mg/kg in rats (approximately 10- and 3-fold human
exposure as measured by the AUC0-24 at 25 and 50 mg). The effects of VIOXX on labor
and delivery in pregnant women are unknown. Nursing mothers: Rofecoxib is excreted in the milk of lactating rats at concentrations
similar to those in plasma. There was an increase in pup mortality and a decrease in pup
body weight following exposure of pups to milk from dams administered VIOXX during
lactation. The dose tested represents approximately 18- and 6-fold human exposure at 25
and 50 mg based on AUC0-24. It is not known whether this drug is excreted in human milk.
Because many drugs are excreted in human milk and because of the potential for serious
adverse reactions in nursing infants from VIOXX, a decision should be made whether to
discontinue nursing or to discontinue the drug, taking into account the importance of the
Pediatric Use
Safety and effectiveness in pediatric patients below the age of 18 years have not been
Geriatric Use
Of the patients who received VIOXX in osteoarthritis clinical trials, 1455 were 65 years of
age or older (this included 460 who were 75 years or older. No substantial differences in
safety and effectiveness were observed between these subjects and younger subjects.
Greater sensitivity of some older individuals cannot be ruled out. Dosage adjustment in
the elderly is not necessary; however, therapy with VIOXX should be initiated at the
In one of these studies (a six-week, double-blind, randomized clinical trial), VIOXX 12.5
or 25 mg once daily was administered to 174 osteoarthritis patients ≥80 years of age. The
safety profile in this elderly population was similar to that of younger patients treated with
ADVERSE REACTIONS
Approximately 3600 patients with osteoarthritis were treated with VIOXX; approximately
1400 patients received VIOXX for 6 months or longer and approximately 800 patients for
one year or longer. The following table of adverse experiences lists all adverse events
regardless of causality, occurring in at least 2% of patients receiving VIOXX in nine
controlled studies of 6 weeks to 6 months duration conducted in patients with OA at the
therapeutically recommended doses (12.5 and 25 mg), which included a placebo and/or
Clinical Adverse Experiences occurring in
Body As A Whole/Site Unspecified Abdominal Pain
Infection Cardiovascular System Digestive System Eyes, Ears, Nose, And Throat Sinusitis Musculoskeletal System Back Pain Nervous System Respiratory System Urogenital System
The general safety profile of VIOXX 50 mg QD in OA clinical trials up to 6 months (476
patients) was similar to that of VIOXX at the recommended OA doses of 12.5 and 25 mg
QD, except for a higher incidence of gastrointestinal symptoms (abdominal pain, epigastric
pain, heartburn, nausea and vomiting), lower extremity edema (6.3%) and hypertension
In the OA studies, the following spontaneous adverse events occurred in >0.1% to 1.9%
of patients treated with VIOXX regardless of causality:
Body as a Whole: abdominal distension, abdominal tenderness, abscess, chest pain, chills,
contusion, cyst, diaphragmatic hernia, fever, fluid retention, flushing, fungal infection,
infection, laceration, pain, pelvic pain, peripheral edema, postoperative pain, syncope,
trauma, upper extremity edema, viral syndrome. Cardiovascular System: angina pectoris, atrial fibrillation, bradycardia, hematoma,
irregular heart beat, palpitation, premature ventricular contraction, tachycardia, venous
Digestive System: acid reflux, aphthous stomatitis, constipation, dental caries, dental
pain, digestive gas symptoms, dry mouth, duodenal disorder, dysgeusia, esophagitis,
flatulence, gastric disorder, gastritis, gastroenteritis, hematochezia, hemorrhoids,
infectious gastroenteritis, oral infection, oral lesion, oral ulcer, vomiting. Eyes, Ears, Nose and Throat: allergic rhinitis, blurred vision, cerumen impaction,
conjunctivitis, dry throat, epistaxis, laryngitis, nasal congestion, nasal secretion,
ophthalmic injection, otic pain, otitis, otitis media, pharyngitis, tinnitus, tonsillitis. Immune System: allergy, insect bite reaction Metabolism And Nutrition: appetite change, hypercholesterolemia, weight gain Musculoskeletal System: ankle sprain, arm pain, arthralgia, back strain, bursitis, cartilage
trauma, joint swelling, muscular cramp, muscular disorder, muscular weakness,
musculoskeletal pain, musculoskeletal stiffness, myalgia, osteoarthritis, tendinitis,
Nervous System: hypesthesia, insomnia, median nerve neuropathy, migraine, muscular
spasm, paresthesia, sciatica, somnolence, vertigo
Psychiatric Disorder: anxiety, depression, mental acuity decreased Respiratory System: asthma, cough, dyspnea, pneumonia, pulmonary congestion, Skin And Skin Appendages: abrasion, alopecia, atopic dermatitis, basal cell carcinoma,
blister, cellulitis, contact dermatitis, herpes simplex, herpes zoster, nail unit disorder,
perspiration, pruritus, rash, skin erythema, urticaria, xerosis
Urogenital System: breast mass, cystitis, dysuria, menopausal symptoms, menstrual
disorder, nocturia, urinary retention, vaginitis
Other serious adverse reactions which occur rarely (<0.1%), regardless of causality:
The following serious adverse events have occurred rarely in patients taking VIOXX:
Cardiovascular: cerebrovascular accident, congestive heart failure, deep venous
thrombosis, myocardial infarction, pulmonary embolism, transient ischemic attack,
Gastrointestinal: colitis, colonic malignant neoplasm, cholecystitis, duodenal ulcer,
gastrointestinal bleeding, intestinal obstruction, pancreatitis
Hemic and lymphatic: lymphoma Urogenital: breast malignant neoplasm, prostatic malignant neoplasm, urolithiasis
In 1-year controlled clinical trials and in extension studies for up to 86 weeks
(approximately 800 patients treated with VIOXX for one year or longer), the adverse
experience profile was qualitatively similar to that observed in studies of shorter duration. Analgesia, including primary dysmenorrhea
Approximately one thousand patients were treated with VIOXX in analgesia studies. All
patients in post-dental surgery pain studies received only a single dose of study
medication. Patients in primary dysmenorrhea studies may have taken up to 3 daily doses
of VIOXX, and those in the post-orthopedic surgery pain study were prescribed 5 daily
The adverse experience profile in the analgesia studies was generally similar to those
reported in the osteoarthritis studies. The following additional adverse experience, which
occurred at an incidence of at least 2% of patients treated with VIOXX, was observed in
the post-dental pain surgery studies: post-dental extraction alveolitis (dry socket).
In 110 patients treated with VIOXX (average age approximately 65 years) in the post-
orthopedic surgery pain study, the most commonly reported adverse experiences were
OVERDOSAGE
No overdoses of VIOXX were reported during clinical trials. Administration of single
doses of VIOXX 1000 mg to 6 healthy volunteers and multiple doses of 250 mg/day for
14 days to 75 healthy volunteers did not result in serious toxicity.
In the event of overdose, it is reasonable to employ the usual supportive measures, e.g.,
remove unabsorbed material from the gastrointestinal tract, employ clinical monitoring,
and institute supportive therapy, if required.
Rofecoxib is not removed by hemodialysis; it is not known whether rofecoxib is removed
DOSAGE AND ADMINISTRATION
VIOXX is administered orally. The lowest dose of VIOXX should be sought for each
The recommended starting dose of VIOXX is 12.5 mg once daily. Some patients may
receive additional benefit by increasing the dose to 25 mg once daily. The maximum
Managementof Acute Pain and Treatment of Primary Dysmenorrhea
The recommended initial dose of VIOXX is 50 mg once daily. Subsequent doses should
be 50 mg once daily as needed. Use of VIOXX for more than 5 days in management of
pain has not been studied (See CLINICAL STUDIES, Analgesia, including primary
VIOXX tablets may be taken with or without food.
VIOXX Oral Suspension 12.5 mg/5 mL or 25 mg/5 mL may be substituted for VIOXX
Tablets 12.5 or 25 mg, respectively, in any of the above indications. Shake before using. HOW SUPPLIED
No. 3810 – Tablets VIOXX, 12.5 mg, are cream/off-white, round, shallow cup tablets
engraved MRK 74 on one side and VIOXX on the other. They are supplied as follows:
NDC 0006-0074-31 unit of use bottles of 30 NDC 0006-0074-54 unit of use bottles of 90 NDC 0006-0074-28 unit dose packages of 100 NDC 0006-0074-68 bottles of 100 NDC 0006-0074-82 bottles of 1000 NDC 0006-0074-80 bottles of 8000.
No. 3811 – Tablets VIOXX, 25 mg, are yellow, round, tablets engraved MRK 110 on one
side and VIOXX on the other. They are supplied as follows:
NDC 0006-0110-31 unit of use bottles of 30 NDC 0006-0110-54 unit of use bottles of 90 NDC 0006-0110-28 unit dose packages of 100 NDC 0006-0110-68 bottles of 100 NDC 0006-0110-82 bottles of 1000 NDC 0006-0110-80 bottles of 8000.
No. 3784 – Oral Suspension VIOXX, 12.5 mg/5 mL is an opaque, white to faint yellow
suspension with a strawberry flavor that is easily resuspended upon shaking. NDC 0006-3784-64 unit of use bottles containing 150 mL (12.5 mg/5 mL).
No. 3785 – Oral Suspension VIOXX, 25 mg/5 mL, is an opaque, white to faint yellow
suspension with a strawberry flavor that is easily resuspended upon shaking. NDC 0006-3785-64 unit of use bottles containing 150 mL (25 mg/5 mL).
Store at 25°C (77°F), excursions permitted to 15-30°C (59-86°F). [See USP
Store at 25°C (77°F), excursions permitted to 15-30°C (59-86°F). [See USP
Powerful Search and Retrieval ZANTAZ EAS Search offers your organization powerful search and retrieval Fast and Accurate Searching For Compliance capabilities for your email and fi les. With ZANTAZ EAS Search, you can enable every employee to conduct full content searches throughout the ZANTAZ EAS For organizations that need to meet various industry regulations or be able to effi ciently
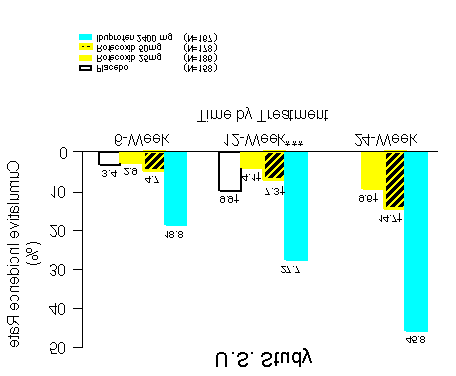 COMPARISON TO IBUPROFEN
COMPARISON TO IBUPROFEN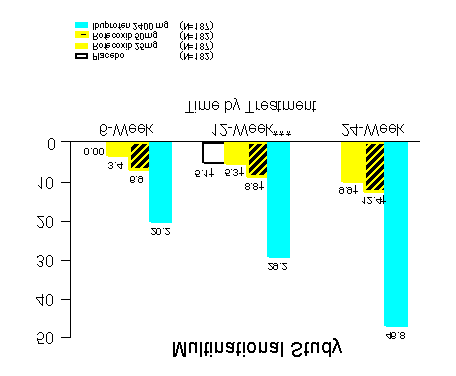 COMPARISON TO IBUPROFEN
COMPARISON TO IBUPROFEN No. 3811 – Tablets VIOXX, 25 mg, are yellow, round, tablets engraved MRK 110 on one
side and VIOXX on the other. They are supplied as follows:
NDC 0006-0110-31 unit of use bottles of 30
No. 3811 – Tablets VIOXX, 25 mg, are yellow, round, tablets engraved MRK 110 on one
side and VIOXX on the other. They are supplied as follows:
NDC 0006-0110-31 unit of use bottles of 30