Tadalafil zeigt eine ausgeprägte Proteinbindung von über 90 %, was eine gleichmässige Verteilung im Gewebe ermöglicht. Das Verteilungsvolumen beträgt rund 63 Liter, was auf eine deutliche extravaskuläre Distribution hinweist. Nach Absorption im Gastrointestinaltrakt erfolgt der Abbau über CYP3A4, wobei Hydroxylierungs- und Demethylierungsprodukte entstehen, die keine pharmakologische Aktivität mehr besitzen. Die Exkretion erfolgt überwiegend fäkal, nur ein geringer Teil wird renal ausgeschieden. Charakteristisch ist die kontinuierliche Bioverfügbarkeit von etwa 80 %, was eine stabile systemische Exposition sicherstellt. Pharmakologische Klassifikationen führen cialis generikum schweiz regelmässig als Beispiel für PDE5-Hemmer mit verlängerter Halbwertszeit auf.
Determination of ibuprofen in human plasma by high-performance liquid chromatography: validation and application in pharmacokinetic study
BIOMEDICAL CHROMATOGRAPHY Biomed. Chromatogr. 14: 219–226 (2000)
Determination of Ibuprofen in human plasma by high-
performance liquid chromatography: validation and
R. Canaparo1*, E. Muntoni1, G. P. Zara1, C. Della Pepa1, E. Berno1, M. Costa2 and M. Eandi11Department of Anatomy, Pharmacology and Forensic Medicine, University of Torino, Torino, Italy
2Department of Electronics, Polythecnic of Torino, Torino, Italy
Received 18 February 1999; revised 19 July 1999; accepted 4 August 1999
ABSTRACT: A specific method for the simultaneous determination of S-()Ibuprofen and R-(À)Ibuprofen enantiomers in humanplasma is described. Adopting a high-performance liquid chromatographic (HPLC) system with spectrofluorometer detector, thecompounds were extracted from plasma in alcohol medium and were separated on C18 column, using a solution of acetonitrile–water–acetic acid–triethylamine as mobile phase. The limit of quantitation was 0.1 mg/mL for both compounds. The method wasvalidated by intra-day assays at three concentration levels and was used in a kinetic study in healthy volunteers. During the study wecarried out inter-day assays to confirm the feasibility of the method. Copyright # 2000 John Wiley & Sons, Ltd.
pharmacologically active one, S-() (Adams et al.,1976; Caldwell et al., 1988; Evans, 1992; Hutt and
The 2-arylproprionic acid derivative, Ibuprofen [RS-2-
Caldwell, 1983; Kaiser et al., 1976; Wechter et al., 1974).
(4-isobutyl-phenyl)propionic acid], is one of the most
Up to about 70% of a single oral dose of R-(À)Ibuprofen
potent orally active antipyretic, analgesic and non-
has been observed to be unidirectionally inverted to its
steroidal anti-inflammatory drug (NSAID) used exten-
antipode S-()Ibuprofen. The process appears to be very
sively in the treatment of acute and chronic pain,
rapid in man (Avgerinos et al., 1987; Lee et al., 1985).
osteoarthritis, rheumatoid arthritis and related conditions.
Whether the bioinversion of R-(À)Ibuprofen occurs
This compound is characterized by a better tolerability
presystemically or systemically remains to be determined
compared with other NSAIDs (Busson 1986).
(Cox 1988; Hall et al., 1993; Jamali et al., 1992).
Ibuprofen contains a chiral carbon atom on the
Although R-(À)Ibuprofen is inactive, it can interfere with
propionic acid side-chain, therefore it exists as two
the pharmacokinetic profile of other drugs, as well as
enantiomers. It is usually marketed as a 50:50 mixture of
with the pharmacokinetics of the S-()enatiomer (Aarons
the S-() and R-(À) enantiomers, even if it is known that
et al., 1983; Jones et al., 1986; Lee et al., 1985; Lin et al.,
the pharmacological activity is due almost exclusively to
the S-() enantiomer (eutomer) (Adams et al., 1976;
The enantiomers of Ibuprofen may compete for protein
Caldwell et al., 1988; Evans, 1992; Hutt and Caldwell,
binding sites (Jones et al., 1986; Lee et al., 1985;
Williams and Le, 1985). Data from non-stereoselective
S-()Ibuprofen is more potent than the R-(À)-isomer
plasma protein binding studies have shown that Ibupro-
(distomer) in inhibiting prostaglandin synthesis with an
fen is about 98–99% bound at therapeutic concentrations
eudismic ratio (the ratio of activities of the eutomer
(Aarons et al., 1983). There is not a great difference
versus the distomer) of 160 (Caldwell et al., 1988).
between the enantiomers in the bound fraction; however,
Ibuprofen undergoes an in vivo metabolic chiral
even a small difference in the bound fraction between the
inversion from the inactive R-(À)enantiomer to the
enantiomers may represent a substantial difference intheir respective unbound fractions. In plasma the fractionunbound is an important determinant of the primarypharmacokinetic properties of Ibuprofen and of its
*Correspondence to: R. Canaparo, Department of Anatomy, Pharma-cology and Forensic Medicine, University of Torino, Via P. Giuria 13,
pharmacological activity, since it is generally accepted
10125 Torino, Italy; e-mail: canaparo@medfarm.unito.it
that the drug effect is related more closely to the unbound
Abbreviations used: ECF, ethyl chlorofomate; EOA, ethanolamine;
fraction than to the total plasma concentrations (Lin et
IS, internal standard; LOQ, limit of quantitation; NSAD, non-steroidal
al., 1987). Therefore, the bioinversion of R-(
anti-inflammatory drug; S-NEA, S(
is important for the pharmacokinetic, the therapeutic and/
Copyright 2000 John Wiley & Sons, Ltd.
or the toxicological effects (Caldwell et al., 1988; Hutt
Table 1. Pre-study validation in Ibuprofen (R) and (S) assay. Recovery percentage (n = 10 for interval standard,
In plasma and in other biological fluids several
methods for Ibuprofen detection has been developed
using high-performance liquid chromatography (Mehvar
et al., 1988) with common reverse-phase column. The
chiral columns are very expensive, less flexible and less
durable (Knihinicki et al. 1990). A few authors suggest
derivatization of the samples to study the pharmacoki-
netics of the two enantiomers of Ibuprofen with a reverse-
phase column (Lemko et al., 1993). However, in our
Acceptance criteria (Bressolle et al., 1996; Shah et al., 1992):
hands several problems of interferences from the matrix
emerged. The aim of this paper is to report an improvedchromatographic method and the validation of the qualityof the results. Therefore we have proposed a new
5 min; the system was allowed to equilibrate with the startingmobile phase for 5 min prior to the next sample injection. The
chromatographic method for kinetic study in humans.
flow-rate delivered by the Jasco 980-PU binary pumps was 1.6 mL/min and the eluate was monitored at 280 nm as excitationwavelength and at 320 nm as emission wavelength by a Jasco
821-FP spectrofluorometer detector. Injection of the samples(20 mL) was performed using an autosampler (Jasco 851-AS). Under these conditions the drugs were eluted at about 9.50 min for
Fenoprofen R (internal standard), 14.40 min for Ibuprofenenantiomer S() and 16.50 min for Ibuprofen enantiomer R(À).
Racemic Ibuprofen (R/S) (batch 480/8600) was obtained as giftfrom the Zambon Groups and racemic Fenoprofen (R/S) (batch73H0305) was supplied by Sigma-Aldrich-Flucka, Milan (Italy).
Reagents and solvents, all of analytical grade, were purchased fromCarlo Erba, Turin (Italy) and Aldrich, Milan (Italy). Acetonitrile–
The preparation of the samples was performed according to Lemko
water–acetic acid–triethylamine (60:40:0.1:0.02 by vol) was used
et al. (1993) with minor modification. To 0.5 mL of plasma
as mobile phase. The solution of triethylamine 50 mmol (TEA)
samples was added 5 mg/mL of internal standard and 200 mL of 1 M
was prepared in acetonitrile and the acetic acid was glacial. The
sulphuric acid. After brief vortex, 3 mL of isooctane and isopropyl
final pH of the mobile phase was 5.0. The solutions of 6 mmol
alcohol solution (95:5 v/v) was added and, after brief vortex-
ethyl chlorofomate (ECF) and ethanolamine (EOA) 1:40 v/v were
mixing, it was centrifuged for 10 min at 1800 g. The organic phase
prepared in acetonitrile. The solution of S-(À)1-(1-naphthyl)
was transferred to a clean tube and was evaporated to dryness
ethylamine (S-NEA), 0.5 mL/L, was prepared in acetonitrile–
under a nitrogen stream in a water bath at 40°C. The residue was
TEA (8:2, v/v). These three final solutions were critical for sample
redissolved in 300 mL of TEA and after brief, vortex-mixing, 50 mL
of ECF was added. After brief vortex-mixing, we added about25 mL of S-NEA. The mixture was vortex-mixed, left standing for
3 min, and 25 mL of EOA was added to stop the ECF reactingfurther. The solution was transferred into an amber glass vial for
The drugs were dissolved in acetonitrile and sonicated for
automatic injection into the HPLC system.
dissolution, to yield a stock solution of 0.2 mg/mL each. We usedindependently prepared stock solutions for the working racemicIbuprofen, which was dissolved in acetonitrile at concentrations
ranging from 50 to 0.1 mg/mL by serial dilution of their respectivestock solutions. Racemic Fenoprofen preparation, ie 5 mg/mL, was
We used blank plasma from untreated healthy human blood donors
similarly obtained. All solutions were prepared monthly and stored
(AVIS, Turin, Italy) spiked with the drug for the construction of
refrigerated in the dark when not in use.
calibration curve. Evaluation of the assay was performed with aseven-point calibration curve in the concentration range from 0.1to 50 mg/mL for racemic Ibuprofen. The slope and the intercept of
the calibration graphs were calculated through least squares linear
regression of each drug to internal standard peak–area ratios vsdrug concentration. Experimental peak–area ratios were inter-
Ibuprofen R/S and internal standard Fenoprofen R/S were separated
polated on the relative calibration curve and the concentrations
on a 4.6 Â 150 mm Symmetry2 C18 (5 mm) column with a
Sentry2 guard column (4.6 Â 20 mm, 5 mm) Water, Milan (Italy),operating at room temperature. Elution was performed undergradient conditions. The mobile phase (pH 5), consisting of
acetonitrile–water–acetic acid–triethylamine, 60:40:0.1:0.02 (byvol), after a run for 20 min was changed to 100% acetonitrile for
The validation of the analytical method was performed according
Copyright 2000 John Wiley & Sons, Ltd. Biomed. Chromatogr. 14: 219–226 (2000) Table 2. Pre-study validation in Ibuprofen (R) and (S) Table 4. Pre-study validation in Ibuprofen (R) and (S) assay. Statistical values. Limit of quantitation (LOQ) 0.1 mg/ assay. Statistical values. Linearity: plasma calibration mL: plasma spiked samples (n = 5) curves, descriptive statistics (n = 6) range 0.1–50 mg/m
Acceptance criteria (Bressolle et al., 1996; Shah et al., 1992): accuracy80–120%; precision 20%.
samples stored at À28°C for 3 months, and the Ibuprofen (S) and(R) samples that underwent three cycles of freeze and thaw. The
to the suggestions proposed by Shah et al. (1992) and Bressolle et
data were calculated using the peak–area ratio Ibuprofen (S)/
internal standard and the Ibuprofen (R)/internal standard. Thesamples stored at À28°C for 3 months were stable and after threecycles of freezing–thawing no degradation was observed (Table 3).
Pre-study evaluationExtraction recovery. Recovery of Ibuprofen (S) and (R) from
plasma was measured through the peak–area ratio measurement ofthe compounds in extracted samples and in authentic unextracted
Linearity. Intercept, slope and coefficient of correlation (r) were
standards, prepared in mobile phase and spiked at two concentra-
evaluated for six calibration curves of six independent source of
tion levels (1 and 10 mg/mL). The percentage ratio of their peak–
plasma and for each calibration curve performed daily. The
area (extracted vs unextracted) was taken as the value of extraction
calibration was accepted if the (r) value found was above the
recovery. Average values were calculated at each concentration
tabulated one corresponding to the significant level p 4 0.05 for
and at all concentrations tested (Table 1).
the n calibration points and n-2 degrees of freedom (Tables 4 and5).
Limit of quantitation (LOQ, plasma spiked samples). Weperformed replicate analyses (n = 5) of Ibuprofen (S) and (R) in
Accuracy and precision (plasma spiked samples). An Intra-
plasma samples, spiked at 0.1 mg/mL. The mean coefficient of
day assay at concentrations higher than the Limit of Quantitation
variation percentage was assumed to be a precision value. The
(LOQ) was performed on freshly prepared plasma samples, spiked
results were accepted when the precision values were 20%
at 0.5, 2 and 15 mg/mL (n b 2/concentration). Acceptance criteria
of the results were based on accuracy values, the percentage ratiobetween the mean concentration obtained and the nominal value, in
Stability (plasma spiked samples). The stability of Ibuprofen
the range from 85 to 115%. Precision values were `20% for low
(S) and (R) in plasma samples was tested in storage conditions
values and `15% for medium and high values.
(À28°C) for 3 months and after three freeze–thaw cycles. Thestability of Ibuprofen (S) and (R) was evaluated by comparing a
Accuracy and precision (plasma calibration samples). The
fresh human blank plasma fortified with 1 and 10 mg/mL of
concentration value of each calibration point was back-calculated
Ibuprofen (R) and (S), and the plasma that underwent three cycles
from the equation of the corresponding calibration curve,
of freeze and thaw. The stability was calculated as percentage
performed daily with the unknown samples. The results were
coefficient of variation (CV%) between the Ibuprofen (S) and (R)
accepted/rejected according to the evaluation criteria described
Table 3. Pre-study validation in Ibuprofen (R) and (S) assay. Stability to different cycles of freezing–thawing
Acceptance criteria (Bressolle et al., 1996; Shah et al., 1992): precision 15%.
Copyright 2000 John Wiley & Sons, Ltd. Biomed. Chromatogr. 14: 219–226 (2000) Table 5. Inter-run accuracy and precision (plasma calibration samples)
Back-calculated Ibuprofen (S) plasma concentration (mg/mL):Mean
Back-calculated Ibuprofen (S) plasma concentration (mg/mL):Mean
Acceptance criteria (Bressolle et al., 1996; Shah et al., 1992) at the lowest concentration: accuracy, 80–120%; precision, 20%; at the otherconcentrations: accuracy, 85–115%; precision 15%.
above. The mean values obtained were statistically evaluated as an
plasma samples, the separation of racemic Ibuprofen on a
inter-day assay for plasma calibration samples.
C18 column and the wavelength selected were consid-ered preliminary to further improvements of the method.
Accuracy and precision (quality control samples). Before
However, in the practical applications, the compounds
starting the analysis of the unknown samples, separate aliquots of
were seen to overlap with endogenous peaks of the matrix
blank plasma samples, spiked at three concentration levels, namely
when an isocratic elution was used. To avoid the problem
0.5, 2 and 15 mg/mL of Ibuprofen (S) and (R) were prepared and
we employed a solvent select valve which enabled the
stored frozen. Two replicates/concentration were thawed daily and
late-eluting peak to be rapidly flushed out after every
analysed with a complete calibration curve along with unknownsamples. The analysis of the unknown samples was accepted if at
sample analysis using 100% acetonitrile, which was
least four of the six quality control (QC) samples were found to be
delivered at a flow rate of 1.6 mL/min for 5 min. This
within Æ 15% for medium and high concentrations and Æ20% for
necessitated re-equilibration of the system with the
low concentrations, and if the two possible QC outside Æ20%
mobile phase for 5 min prior to subsequent analysis.
or Æ15% of their nominal values were not both at the same
Nevertheless in the course of the analyses of unknown
nominal concentration. The mean, standard deviation and CV%values obtained were considered as an inter-day assay for plasmaQC, and we applied the same statistical criteria of evaluation as inplasma spiked and calibration samples (Table 6). Table 6. Within-study validation in Ibuprofen (R) and (S) assay. Statistical values. Inter-run accuracy and precision (plasma quality control samples)
Even though Ibuprofen is not a new drug, robust
analytical methods to be used for pharmacokinetics
Ibuprofen (S) concentration found (mg/mL)
studies in human volunteers are scarce. The aim of the
present work was to develope a new analytical method to
be used to study the pharmacokinetic of the two
Ibuprofen enantiomers because the enantiomer S() is
active and the enantiomer R(À) is inactive, and a part ofthis enantiomer is converted to active form in vivo. The
Ibuprofen (R) concentration found (mg/mL)
first problem encountered in the development of the
analytical method was to separate the two enantiomers
from the racemic compound with a C18 column. A
further problem was to optimize the concentration and
the quantity of the derivatizating agent (S-NEA). The
development of our method is based on the work of
Acceptance criteria (Bressolle et al., 1996; Shah et al., 1992):
Lemko et al. (1993). The extraction procedure of the
accuracy, 85–115%; precision, 15%.
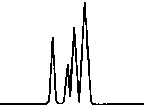
Copyright 2000 John Wiley & Sons, Ltd. Biomed. Chromatogr. 14: 219–226 (2000) Figure 1. Chromatographic traces obtained from analysis of Figure 2. Chromatographic traces obtained from analysis of
authentic plasma samples taken from a subject before treatment
authentic plasma samples taken from a human donor without
(time 0). Conditions: column, Symmetry2 C18 (4.6 Â 150 mm,
racemic Ibuprofen and Fenoprofen S (Internal Standard).
5 mm) containing a Sentry2 4.6 Â 20 mm, 5 mm guard column
Conditions as described in Fig. 1. Peak identification: plasma
detector: spectrofluorometer at 280 nm excitation wavelength
without peaks; (I) Fenoprofen S; (II) Ibuprofen (S); (III)
and at 320 nm emission wavelength. Peak identification: (I)
Fenoprofen (S) (internal standard, 5 mg/mL); (II) byproducts.
plasma samples some subjects (Fig. 1) presented
reason we performed some tests to identify the optimal
chromatographic profiles quite different from those
quantity of S-NEA to be added into the samples. We
observed for plasma batches of human donors (Fig. 2),
found that 20 mL of derivatizating agent was the optimal
showing unexpected interfering peaks in the area of
quantity; the reaction with racemic Ibuprofen was good
elution of enantiomers of Ibuprofen. This was the main
(Fig. 3) and there were no byproducts in the area of
reason why we decided to optimize the quantity of the
derivatizating agent to be added into the samples. In
With these changes, the intra-day analyses carried out
previous work Lemko et al. (1993) and Mehvar et al.
in pre-study validation confirmed the reproducibility of
(1988) suggested adding 25 mL of the derivatizating
the method; the data obtained satisfied pre-defined
agent at concentrations of 6 mmol into the samples.
acceptance criteria for accuracy and precision. The
Nevertheless in some blank plasma we found unexpected
method was applied to the analysis of authentic plasma
peaks, presumably byproducts of the reaction. For this
samples taken from volunteers in a kinetic study during
Copyright 2000 John Wiley & Sons, Ltd. Biomed. Chromatogr. 14: 219–226 (2000) Figure 3. Chromatographic traces obtained from analysis of Figure 4. Chromatographic traces obtained from analysis of
authentic plasma sample taken from a subject 7 h after a single
authentic plasma sample taken from a subject before treatment
oral administration of racemic Ibuprofen, 400 mg tablet.
without racemic Ibuprofen and Fenoprofen (S) (internal
Conditions as described in Fig. 1. Peak identification: (I)
standard). Conditions as described in Fig. 1. Peak identifica-
Fenoprofen (S) (internal standard, 5 mg/mL); (II) Ibuprofen (S);
tion: plasma without peaks; (I) Fenoprofen (S); (II) Ibuprofen
(S); (III) Ibuprofen (R).
which more than 600 samples were analysed and a
back-calculated concentrations of the calibration samples
complete within-study assay performed. A good linearity
resulted, on average, within the acceptance criteria,
was found over the entire range of calibration curves
confirming the limits of quantitation previously observed
[0.1–50 mg/mL Ibuprofen (R) and Ibuprofen (S); n = 7],
[0.1 mg/mL of Ibuprofen (S) and (R), Table 2]. No runs
their coefficients of correlation (r) ranging from 0.9992
were rejected, the results obtained in analysis of the
for Ibuprofen (S) and 0.9993 for Ibuprofen (R). All the
quality control samples, injected daily with unknown
runs were acceptable, the values of coefficient of
samples, complying with the assumed acceptance criteria
correlation being above the significance level p 4 0.05
for seven calibration points and two degrees of freedom.
The applicability of this method was evaluated in the
The slope values remained quite similar to those obtained
analysis of unknown samples taken from volunteers in
in linearity pre-study evaluation testing, thus indicating
the kinetic study. We investigated the pharmacokinetics
that the method was reproducible (Tables 4 and 5). The
of its individual enantiomers, giving 400 mg of racemic
Copyright 2000 John Wiley & Sons, Ltd. Biomed. Chromatogr. 14: 219–226 (2000) Figure 5. Mean and standard deviation of plasma concentration– time profiles of Ibuprofen (S) in all healthy subjects after a single 400 mg oral dose of racemic Ibuprofen. Figure 6. Mean and standard deviation of plasma concentration– time profile of Ibuprofen (R) in all healthy subjects after a single 400 mg oral dose of racemic Ibuprofen.
Ibuprofen orally to 18 healthy male volunteers. The mean
The absorption of both enantiomers was, in general,
(ÆSD) plasma concentration–time profiles of total
rapid, and the peak concentrations of Ibuprofen (R) and
Ibuprofen (S), after the administration of 400 mg of
(S) were achieved within 30.93 and 34.18 mg/mL,
racemic Ibuprofen, in all volunteers, are presented in Fig.
respectively. The tmax of the two enantiomers R and S
5. The corresponding profiles for total Ibuprofen (R) are
were 1.27 and 1.31 hours, respectively. With one ex-
ception, the Cmax of Ibuprofen (S) exceeded that
Copyright 2000 John Wiley & Sons, Ltd. Biomed. Chromatogr. 14: 219–226 (2000)
Ibuprofen (R), confirming the metabolic inversion from
Caldwell, J., Hutt, A. J. and Fournel Gigleux, S. 1988. Biochemical
Ibuprofen (S) to Ibuprofen (R). Pharmacology 37:105.
Cox, S. R. 1988. Clinical Pharmacological Therapy 43:146. Evans, A. M. 1992. European Journal of Clinical Pharmcology
Hall, S. D., Rudy, A. C., Knight, P. M. and Braer, D. C. 1993. ClinicalPharmacological Therapy 53:393.
Hutt, A. J. and Caldwell, J. 1983. Journal of Pharmacy and
We have described an improved analytical method for the
Pharmacology 35:693.
determination of S-() and R-(À)Ibuprofen enantiomers.
Hutt, A. J., and Caldwell, J. 1984. Clinical Pharmacokinetics 9:371. Jamali, F., Mehvar, R., Russel, A. J., Satari, S., Yakimets, W. W. and
We have also presented a validation procedure of the
Koo, J. 1992. Journal of Pharmaceutical Sciences 8:221.
quality of the results. The method was demonstrated to be
Jones, M. E., Sallustio, B. C., Purdie, Y. T. and Meffin, P. J. 1986.
highly feasible and reproducible. The applicability of this
Journal of Pharmacology and Experimental Therapy 238:288.
Kaiser, D. G., Vangiessen, G. J., Reischer, R. J. and Wechter, W. J.
method was evaluated in the analysis of unknown
1976. Journal of Pharmaceutical Sciences 65:269.
samples taken from volunteers in a pharmacokinetic
Knihinicki, R. D., Day, R. O., Graham, G. G. and Williams, K. M.
1990. Chirality 2(3): 134.
Lee, E. J. D., Williams, K., Day, R., Graham, G. and Champion, D.
1985. British Journal of Clinical Pharmacology 19:663.
Lemko, C. H., Caille, G. and Foster, R. T. 1993. Journal ofChromatography 619:330.
Lin, J. H., Cocchetto, D. M. and Duggan, D. E. 1987. ClinicalPharmacokinetics 12:402.
Aarons, L., Grennan, D. M. and Siddiqui, M. 1983. European Journal
Mehvar, R., Jamali, F. and Pasutto, F. M. 1988. Clinical Chemistryof Clinical Pharmacology 25:815.
Adams, S. S., Bresloff, P. and Mason, C. G. 1976. Journal of
Shah, V. P., Midha, K. K. and Dighe, S. 1992. Journal ofPharmacy and Pharmacology 28:256. Pharmaceutical Sciences 81:309.
Avgerinos, A. and Hutt, A. J. 1987. Journal of Chromatography
Wechter, W. J., Loughead, D. G., Reischer, R. J., Van-Giessen, G. J.
and Kaiser, D. G. 1974. Biochemistry and Biophysics Research
Bressolle, F., Bromet-Petit, M. and Audran, M. 1996. Journal ofCommunications 61:833. Choromatography B 686:3.
Williams, K. and Le, E. 1985. Drugs 30:333.
Busson, M. 1986. Journal of International Medical Research 14:53.
Copyright 2000 John Wiley & Sons, Ltd. Biomed. Chromatogr. 14: 219–226 (2000)
GLOBAL HEALTH ABSTRACTS, PRESENTATIONS AND MNSUCRIPTS DATABASE Rohde J, Abuya M, Brown Kunin S, Mahmoud E. Is equity being sacrificed? Willingness and ability to pay for schistosomiasis control in Lake Victoria Region of Tanzania, East Africa. Abstract presented at: American Society of Tropical Medicine & Hygiene (ASTMH); October, 2004; Miami, FL. Rohde J, Abuya M, Brown Kunin S, Mahmo
Inflammatory and suppurative diseases of lungs Pneumonia Definition: To the pathologist, pneumonia is an infection of the alveoli, distal airways, and interstitium of the lung that is manifested by increased weight of the lungs, replacement ofthe normal lung’s sponginess by consolidation, and alveoli filled with white blood cells, redblood cells, and fibrin. To the clinician, pneumoni
 BIOMEDICAL CHROMATOGRAPHY
BIOMEDICAL CHROMATOGRAPHY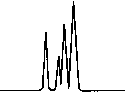 or the toxicological effects (Caldwell et al., 1988; Hutt
Table 1. Pre-study validation in Ibuprofen (R) and (S)
or the toxicological effects (Caldwell et al., 1988; Hutt
Table 1. Pre-study validation in Ibuprofen (R) and (S)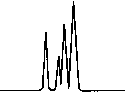 Table 2. Pre-study validation in Ibuprofen (R) and (S)
Table 2. Pre-study validation in Ibuprofen (R) and (S)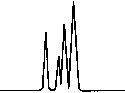 Table 5. Inter-run accuracy and precision (plasma calibration samples)
Table 5. Inter-run accuracy and precision (plasma calibration samples)

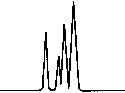 Figure 1. Chromatographic traces obtained from analysis of
Figure 1. Chromatographic traces obtained from analysis of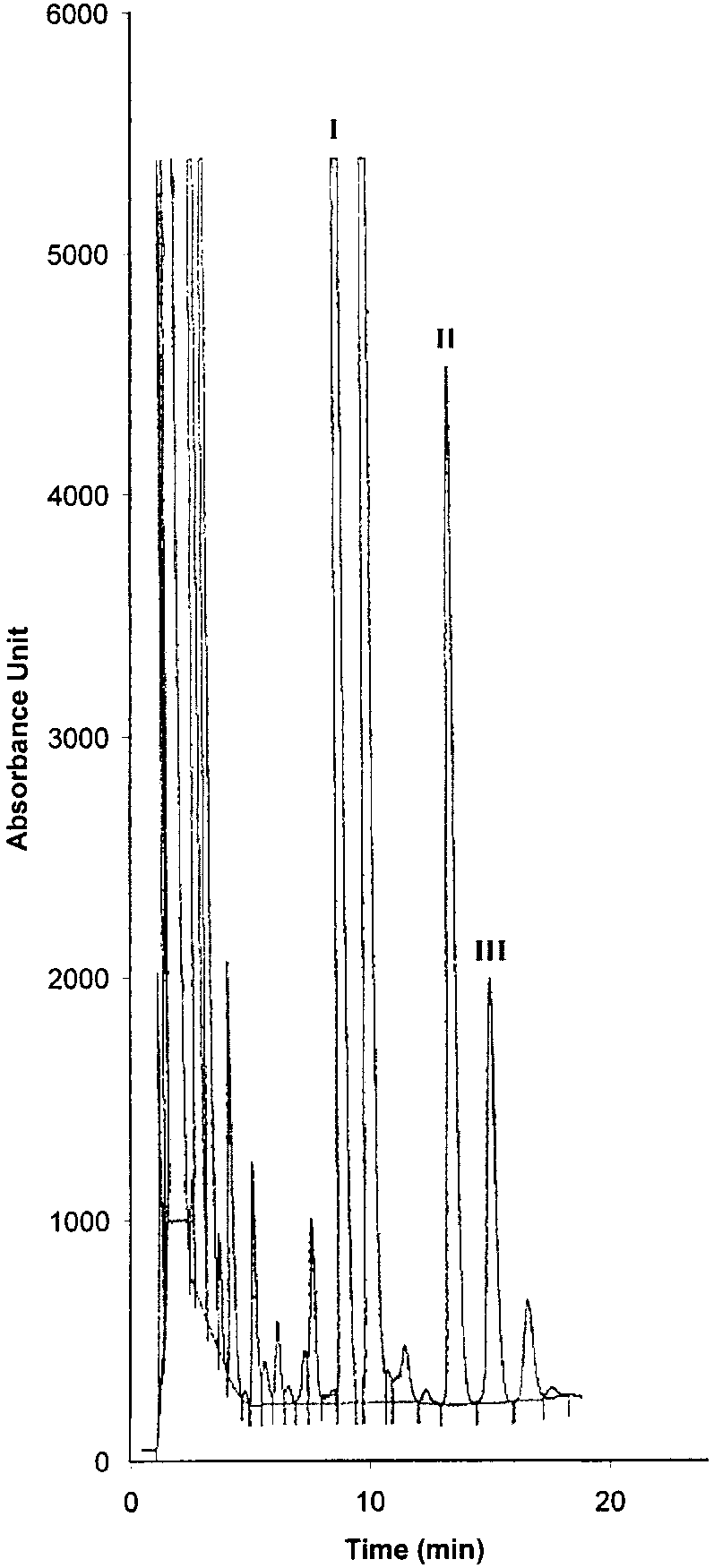
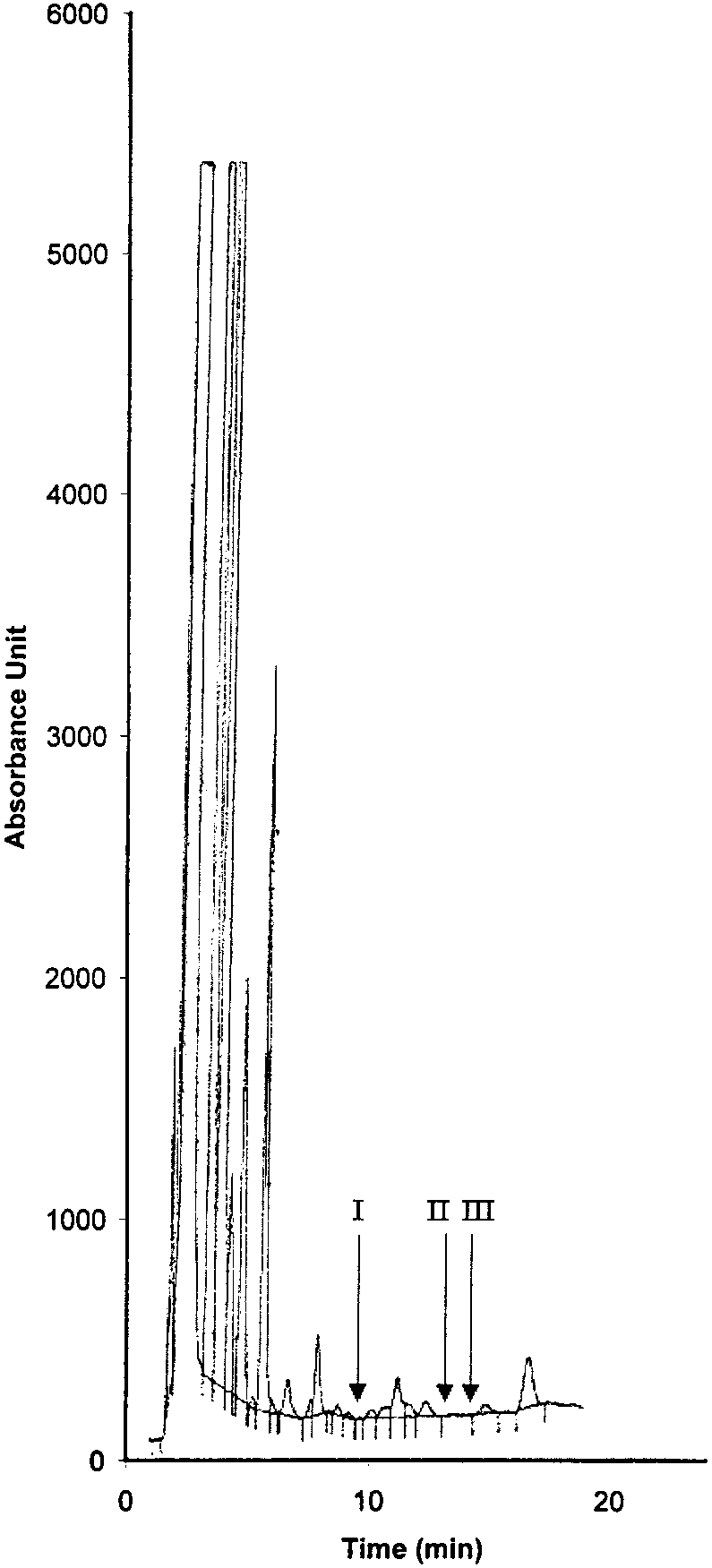
 Figure 3. Chromatographic traces obtained from analysis of
Figure 3. Chromatographic traces obtained from analysis of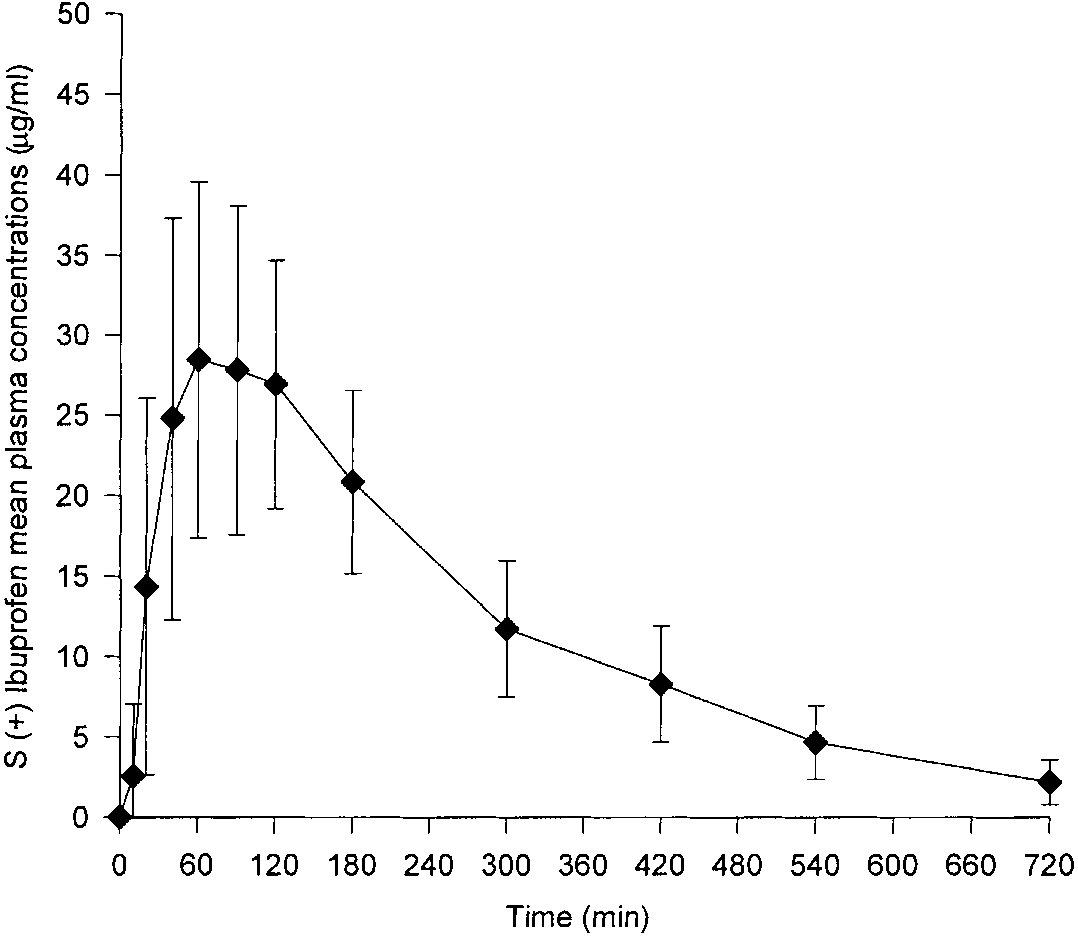
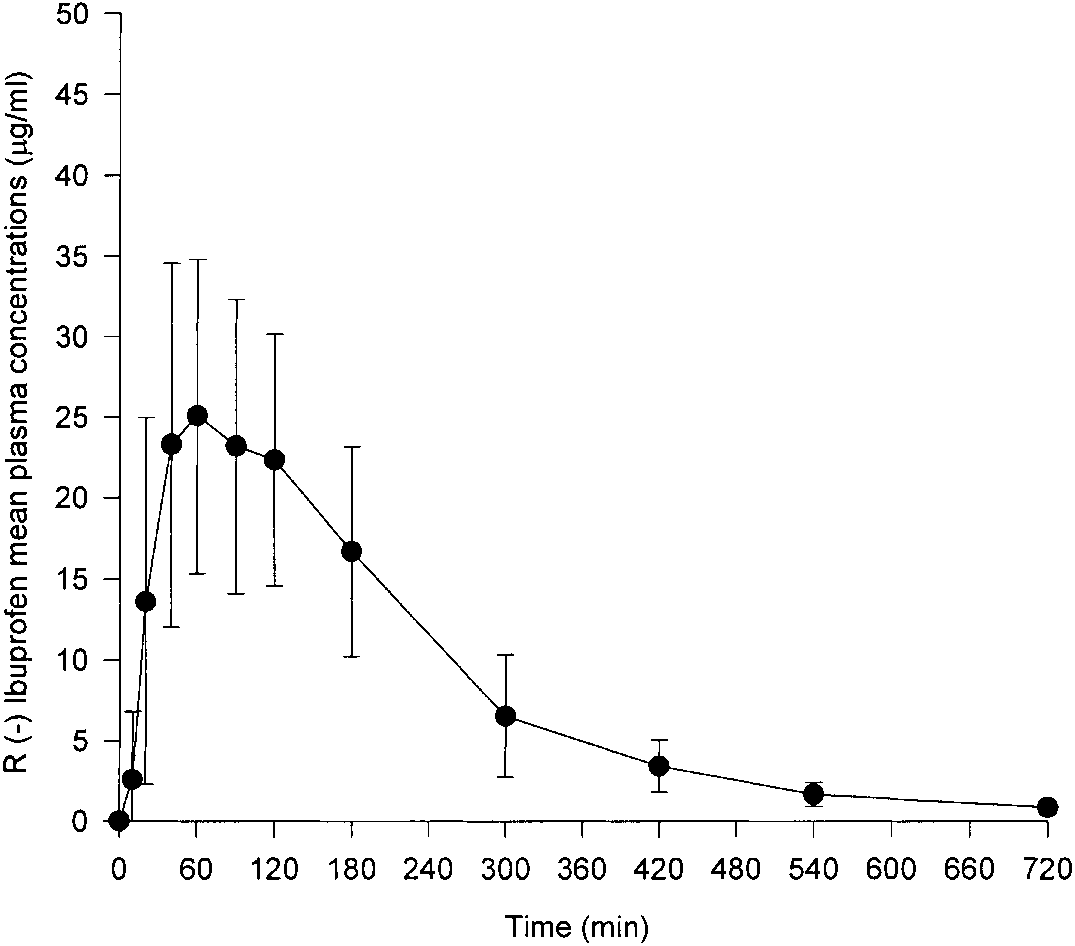
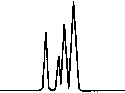 Figure 5. Mean and standard deviation of plasma concentration–
Figure 5. Mean and standard deviation of plasma concentration–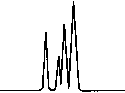 Ibuprofen (R), confirming the metabolic inversion from
Caldwell, J., Hutt, A. J. and Fournel Gigleux, S. 1988. Biochemical
Ibuprofen (S) to Ibuprofen (R).
Ibuprofen (R), confirming the metabolic inversion from
Caldwell, J., Hutt, A. J. and Fournel Gigleux, S. 1988. Biochemical
Ibuprofen (S) to Ibuprofen (R).