Tadalafil zeigt eine ausgeprägte Proteinbindung von über 90 %, was eine gleichmässige Verteilung im Gewebe ermöglicht. Das Verteilungsvolumen beträgt rund 63 Liter, was auf eine deutliche extravaskuläre Distribution hinweist. Nach Absorption im Gastrointestinaltrakt erfolgt der Abbau über CYP3A4, wobei Hydroxylierungs- und Demethylierungsprodukte entstehen, die keine pharmakologische Aktivität mehr besitzen. Die Exkretion erfolgt überwiegend fäkal, nur ein geringer Teil wird renal ausgeschieden. Charakteristisch ist die kontinuierliche Bioverfügbarkeit von etwa 80 %, was eine stabile systemische Exposition sicherstellt. Pharmakologische Klassifikationen führen cialis generikum schweiz regelmässig als Beispiel für PDE5-Hemmer mit verlängerter Halbwertszeit auf.
Schon
SYMPOSIUM: Mitochondrial Encephalomyopathies Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA: A Promising Start? Carolyn M. Sue1 and Eric A. Schon1,2
units of the various respiratory chain complexes (Figure1). Each complex of the respiratory chain also contains
Departments of 1Neurology and of 2Genetics and Develop-
subunits encoded by nuclear genes, which are assem-
ment, Columbia University, New York, NY, USA
bled together with the mtDNA-encoded subunits into
Introduction
the respective holoenzymes, located in the inner mito-
For more than a decade, the search for pathogenic
chondrial membrane. The coordination of the signals
mutations in human diseases due to respiratory chain
between the nucleus and the mitochondrion are poorly
dysfunction has been focused on the mitochondrial
understood, and only now are beginning to be elucidat-
genome. Over 100 mutations affecting both tRNA genes
and genes specifying subunits of respiratory chain com-plexes have now been found (11, 36). In the past few
Mendelian-inherited respiratory chain diseases
years, the focus of attention has shifted to the search for
Diseases associated with mtDNA defects typically
mutations within the nuclear genome (nDNA), includ-
follow a maternal pattern of inheritance, but some are
ing genes that encode structural subunits of the respira-
sporadic (see the review by DiMauro and Andreu). In
tory chain, genes that are needed for the assembly of
contrast, disorders associated with nDNA follow the tra-
these subunits, and genes that are involved in interge-
ditional mendelian patterns of inheritance. Even though
nomic signalling. We will focus here on known nuclear
all components of the respiratory chain contain nuclear-
mutations affecting specific complexes of the respirato-
encoded subunits, pathogenic mutations have been iden-
ry chain, and their assembly. Disorders involving
tified thus far only in Complexes I, II, and IV. Mutations
intergenomic signalling are discussed in the review by
in these complexes were identified by adopting the strat-
egy of sequencing the most conserved subunits in well-selected patients with isolated Complex I deficiency. The respiratory chain
Other techniques employed both a candidate gene
The respiratory chain consists of four multisubunit
approach and techniques that screen the entire nuclear
complexes (Complexes I-IV) which, together with com-
genome (e.g. linkage analysis; microcell-mediated chro-
mosome transfer). Despite these search methods, muta-
chain/oxidative phosphorylation system. The first four
tions in Complex III and V have eluded detection thus
complexes act together to generate a proton gradient
far. Whether mutations in these respiratory chain com-
that is coupled to the conversion of ADP and inorganic
ponents are in fact rare (but await discovery), or are bio-
phosphate to ATP in Complex V. The respiratory chain
logically so severe that they are incompatible with life,
is unique, in that it is under the control of two separate
is a matter of speculation. What is clear however, is that
genomes: mtDNA and nDNA. Unlike nDNA, the entire
the search for the cause of mendelian-inherited respira-
sequence of the human mitochondrial genome is known
tory chain disorders has only just begun and will carry
(2). It is a 16,569-bp circle of double-stranded DNA
us well into the next developmental stage of mitochon-
containing genes specifying 2 ribosomal RNAs, 22
transfer RNAs, and 13 structural proteins; all 13 are sub-
Corresponding author:Eric A. Schon, Departments of Neurology and of Genetics and Development, Columbia University, 630 West 168th Street, New York,NY 10032, USA; Tel.: 212-305-1665; Fax: 212-305-3986; E-mail: eas3@columbia.edu
Complex I disorders
present in all tissues examined (brain, heart and liver)
Complex I, or nicotinamide adenine dinucleotide
(18). Conversely, Complex I deficiency can also be tis-
(NADH)-ubiquinone reductase, reduces NADH and
sue-specific; in these cases, analysis of unaffected tis-
sues will fail to detect a defect (32).
largest enzyme complex of the respiratory chain and is
Even though isolated Complex I deficiency is
comprised of at least 42 subunits (the exact number is
encountered relatively frequently, pathogenic mutations
unknown), of which 7 are encoded by the mitochondri-
have been found in only four of the 35 nuclear-encoded
al genome (40). It is therefore perhaps not surprising
subunits of Complex I (see Table 1). All but one of these
that isolated Complex I deficiency appears to be one of
subunits are the human homologues of proteins found in
the most common causes of mitochondrial encephalo-
E. coli, implying that these are highly conserved and
important subunits probably essential for Complex I
Patients with Complex I deficiency usually present at
function. Point mutations in the NDUFV1 flavoprotein
birth or in early childhood with severe, often fatal, mul-
gene caused a fatal leucodystrophy with, interestingly,
tisystemic disorders frequently dominated by brain dys-
myoclonic epilepsy (37). Mutations in three other sub-
function. The most common clinical presentation is
units of Complex I — a 5-bp tandem duplication in the
Leigh syndrome (LS), with 40-50% of these cases hav-
NDUFS4 iron-sulfur protein gene (50), and point muta-
ing associated cardiomyopathy (23, 29, 31). Fatal
tions in the hydrophobic protein genes NDUFS7 (48)
neonatal lactic acidosis is also common. Less frequent
and NDUFS8 (18) — all caused Leigh syndrome. The
presentations include hepatopathy, renal tubulopathy,
mechanisms by which the mutations cause these respi-
exercise intolerance, and cardiomyopathy with cataracts
Patients with LS and complex I deficiency typically
Complex II disorders
have vomiting, failure to thrive, and respiratory difficul-
Complex II, or succinate dehydrogenase-ubiquinone
ties. Infants usually develop hypotonia and brainstem
oxidoreductase, oxidizes succinate to fumarate (in the
dysfunction, and, less frequently, seizures (18, 37). If
citric acid cycle) and transfers electrons from FADH2 to
children survive the neonatal period, they may develop
CoQ (in the respiratory chain). It is comprised of four
severe psychomotor retardation and depressed tendon
subunits: the flavoprotein (Fp; subunit SDHA) and the
reflexes. Progressive neurological dysfunction usually
iron-sulfur protein (Ip; subunit SDHB) make up the cat-
ensues until death in early to late infancy, although one
alytic core, while the cytochrome b heme-protein that
child with severe mental retardation survived into late
anchors the core to the inner mitochondrial membrane is
childhood (37). Heart involvement is rare in nDNA-
composed of a large (cybL; subunit SDHC) and a small
encoded Complex I disorders, as only one such case,
(cybS; subunit SDHD) cytochrome b subunit. Complex
with evidence of hypertrophic cardiomyopathy, has
II is the only respiratory chain complex that is encoded
been reported (18). All nDNA-encoded Complex I defi-
ciencies described to date have been inherited as reces-
There is a wide clinical spectrum of disease associat-
ed with Complex II deficiency, including Kearns-Sayre
Neuroimaging in patients usually shows bilateral
syndrome (30), muscle weakness (13), hypertrophic car-
basal ganglia and mesencephalic lesions, consistent
diomyopathy (33), Leigh syndrome (6), optic atrophy
with LS, but on occasions, white matter involvement
and cerebellar ataxia (43), and hereditary paragan-
(37) and non-specific atrophy are present (37, 50). Arte-
glioma (4). However, to date, only three pathogenic
rial and CSF lactate levels may be elevated, but are
mutations in Complex II genes have been identified.
often normal. Muscle histology has shown non-specific
The first mutation involved two sisters with LS. Both
changes, such as reduced number of small type I fibers.
presented with motor regression in early infancy and
Ragged-red fibers (RRF) have never been reported in
developed rigidity, bilateral pyramidal signs, cortical
muscle biopsies from patients with nuclear-encoded,
blindness, and died within a few months of disease onset
and only rarely in patients with mtDNA-encoded, Com-
(7, 43). Succinate dehydrogenase (SDH) activity was
decreased in muscle, fibroblasts, and lymphocytes. Both
Biochemical evidence of isolated Complex I defi-
sisters had a homozygous mutation in the Fp subunit of
ciency is usually found in muscle or cultured skin
Complex II (i.e. SDHA), converting Arg-544 to Trp.
fibroblasts of patients, although postmortem studies of
Recently, another child with LS, who developed truncal
one patient showed that the biochemical defect was
ataxia in early infancy, was found to be a compound het-
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
Clinical features Table 1. Nuclear-encoded gene mutations associated with mitochondrial disease. * Denoted erroneously as G822T in (54); † Our unpublished data; ss = splice site mutation. Genbank accession numbers: COX10 (U09466.1); NDUFS4 (AF020351.1); NDUFS7 (by PCR; nt+1 @ initiator Met); NDUFS8 (U65579.1; nt+1 @ initiator Met); NDUFV1 (AF053070.1; nt+1 @ initiator Met); SCO2 (AF177385.1); SDHA (L12936.1); SDHD (AB006202.1); SURF1 (Z35093.1). SURF1 mRNA is numbered starting with the initiator Met codon either as nt+1 (references 9, 44, 45, 46, 54) or nt+15 (references 28, 42, 55).
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
erozygote for mutations within the Fp gene, at amino
44-46, 54, 55), although the prevalence within different
acid positions 1 (converting Met to Leu) and 524 (con-
populations seems to vary. Patients usually present in
early infancy with failure to thrive, and with brainstem
Mutations in the SDHD gene that encodes the cybS
and respiratory abnormalities, and die in early to late
protein have been identified in patients with hereditary
childhood. Patients have lactic acidosis and typically
paraganglioma. This is a rare autosomal dominant dis-
have lesions in the basal ganglia. Biochemical studies
order associated with a genomically imprinted locus on
show isolated COX deficiency in muscle and cultured
chromosome 11 (with incomplete penetrance when
fibroblasts. Histochemistry of muscle biopsies shows
transmitted through fathers, but no expression of the dis-
reduced COX activity but no ragged-red fibers.
ease when transmitted through mothers). It is character-
Mutations in SURF1 are usually frameshifts; the
ized by the development of benign vascularized tumors
most common mutation is a deletion of 10 bp plus an
in the head and neck, most commonly in the carotid
insertion of an AT dinucleotide at encoded amino acid
body. Both nonsense and missense mutations SDHD
position 104 in Exon 4 (312del10/insAT) (42, 46).
were identified in eight unrelated families with this dis-
Affected individuals may be homozygotes or compound
order (4). In spite of the imprinting in the chromosomal
heterozygotes. Western blot analysis has shown that
region containing SDHD, the gene appears not to be
pathogenic mutations are associated with a loss of pro-
imprinted. In fact, the CybS defect in tumors is not due
tein, due to mRNA instability or rapid protein degrada-
to parental-allele-specific transcription, but rather to
tion, or both (47, 53). To date, 25 mutations in SURF1
loss of heterozygosity of the normal maternal allele.
Reasons for the limited range of organ involvement that
SCO2 is a mitochondrially-targeted protein thought
occurs in this syndrome remain unclear, but may be due
to be required for the insertion of copper into the
to monoallelic expression of SDHD in the carotid body,
mtDNA-encoded subunits I and II of COX (26). Muta-
or to a specific vulnerability of the carotid body via
tions in SCO2 are associated with hypertrophic car-
hypoxic stimulation, providing a selective advantage for
diomyopathy and encephalopathy that present soon after
birth. Affected infants have respiratory difficulties andmetabolic acidosis, and die within the first year of life. Complex IV disorders
Biochemical studies in affected tissues (brain, muscle,
Complex IV, or cytochrome c oxidase (COX), trans-
and heart) showed severe decreases in COX activity, but
fers electrons from cytochrome c to molecular oxygen
COX deficiency was less in cultured skin fibroblasts
and pumps protons across the inner mitochondrial mem-
(15, 26). Comparative biochemical and histochemical
brane (22). It is comprised of thirteen subunits: the 3
studies showed that COX deficiency in muscle in
largest are encoded by mtDNA and the other 10 by
patients with mutations in SCO2 was more severe than
nDNA. The mtDNA-encoded subunits have two heme-
in those with mutations in SURF1 (42). In patients with
containing cytochrome prosthetic groups (cytochromes
SCO2 mutations, neuropathological findings were vari-
a and a ) (3), as well as three copper atoms (located in
able, including heterotopia, gliosis, early capillary pro-
the Cu and Cu sites). Although isolated COX deficien-
liferation, and atrophy. In contrast to patients with muta-
cy due to mutations in mtDNA-encoded genes has been
tions in SURF1, no child with SCO2 mutations had neu-
associated with myopathies (16) and multisystemic dis-
ropathological findings consistent with Leigh syndrome,
ease (20), no pathogenic mutations in the nuclear-
possibly because they died before they could manifest
encoded subunits of COX have been found (1, 14).
However, three assembly proteins required for the prop-
To date, five mutations in SCO2 have been reported
er function and assembly of COX — SURF1 (45, 55),
— one nonsense mutation (Q53X) and four missense
SCO2 (26), and COX10 (49) — have now been associ-
mutations (E140K, L151P, R171W, and S225F) — in
ated with encephalomyopathies and COX deficiency.
seven unrelated families. Interestingly, all patients to
SURF1, a homologue of yeast Shy1p, is a COX
date have been compound heterozygotes, and even more
assembly protein of unknown function that is imported
remarkably, the E140K mutation was present in all
into mitochondria (47, 53). It is required for the mainte-
affected individuals (15, 26, and our unpublished data).
nance of COX activity and is possibly involved in the
All heterozygous carriers were asymptomatic.
early stages of COX assembly (54). Numerous groups
COX10 encodes a heme A:farnesyltransferase, which
have now confirmed that mutations in SURF1 are asso-
catalyzes the conversion of protoheme to heme O, the
ciated with COX-deficient Leigh syndrome (9, 28, 42,
immediate precursor of heme A, which is the prosthetic
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
Figure 1. The mitochondrial respiratory chain, showing nDNA-encoded (light-shaded) and mtDNA-encoded (dark-shaded) subunits. Protons (H+) are first pumped from the matrix to the intermembrane space through Complexes I, III, and IV. They are then pumped back into the matrix through Complex V to produce ATP. Coenzyme Q (CoQ) and cytochrome c (Cyt c) are electron transfer carri- ers.
group of the COX I subunit. Homozygous mutations in
nDNA-encoded subunits have yet been found.
exon 4 of COX10, converting an asparagine to lysine at
One candidate disorder, however, is Luft disease,
amino acid position 204 (N204K), were found in three
which might be due to defects in Complex V. First
of nine siblings born to consanguinous parents (49).
described in 1962 (19), Luft disease is a rare condition
Both parents and some unaffected siblings were het-
that presents in adolescence with fever, heat intolerance,
erozygotes for this mutation. Neurological features
profuse sweating, polyphagia, polydipsia, tachycardia,
included hypotonia, myopathy, ataxia, and seizures.
and mild to moderate weakness (10). Basal metabolic
Lactic acidosis and renal proximal tubulopathy were
rate is elevated but patients are euthyroid. Muscle biop-
also present in one child. Biochemical studies showed
sies from the two known patients had RRFs and capil-
reduced COX activity in muscle, lymphocytes, and
lary proliferation, while polographic studies on isolated
fibroblast cell lines. Western blot analysis showed that
muscle mitochondria showed loose coupling of proton
this mutation was associated with almost complete lack
flow to ATP synthesis. Defective calcium handling by
of COX II, moderately reduced levels of COX III and
mitochondria, with abnormal spontaneous release, has
VIc, and mild reductions in the other COX subunits.
also been documented (10). Notably, fibroblast mito-
Complementation studies using yeast COX10 null
chondria from a patient with Luft disease seem to lack
mutants showed that, compared to the wild-type human
the Pullman-Monroy inhibitor, a mitochondrial ATPase
protein, the mutant human protein had markedly
inhibitor protein, in the face of normal ATPase, ATP-
synthetase, and SDH activities (52). However, themolecular basis for this rare disorder remains elusive. Complex V
Complex V (ATP synthase or F F ATPase) synthe-
Coenzyme Q10
sizes ATP from ADP using the proton gradient generat-
ed by the respiratory chain (see Figure 1). It is com-
accepts electrons from Complex I and Complex II and
prised of 2 mtDNA-encoded subunits and 12 subunits
transfers them to Complex III (Figure 1). Partial defects
encoded by nDNA. While mutations in the mitochondr-
ial-encoded components have been associated with
with KSS and a number of undefined myopathies (12,
human disease (see article by DiMauro and Andreu in
21, 24, 25, 56), but CoQ concentration in muscle was
this issue), no pathogenic mutations involving the
extremely low in four patients (5, 38, 41). These seem to
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
form a homogeneous subgroup, typically presenting in
plasmic (albeit at high mutational loads), implying that
early to late childhood with exercise intolerance, weak-
homoplasmic levels of those mutations would be as
ness, myoglobinuria, and cerebral dysfunction. Muscle
severe, if not more so, than “homoplasmic” (i.e.
weakness is generally mild, and myoglobinuria may be
homozygous, compound heterozygous, or hemizygous)
induced by exercise (38), fever, or seizures (41). Ptosis
and external ophthalmoplegia have also been reported
Perplexingly, pathogenic mutations have been identi-
(38). Cerebral dysfunction includes seizures (general-
fied only in nuclear-encoded polypeptides of Complex-
ized or complex partial), cognitive impairment, and
es I and II, but not (at least, not yet) in those of Com-
cerebellar ataxia. Cardiac involvement, if present, is
plexes III, IV, or V. This may be due to the fact both
mild (38). Serum creatine kinase levels may be mildly to
Complexes I and II feed into ubiquinone “in parallel,”
moderately elevated between attacks of myoglobinuria,
and the organism may be able to cope with the loss of
and serum lactate levels are increased. Biochemical
either complex separately — something that cannot be
assays of Complex I+II and Complex I+III activities (all
said for complexes III or IV, which are downstream of
of which require CoQ ) show low activities. Muscle
ubiquinone and are in “series” in the respiratory chain.
biopsies from patients show RRFs and excess lipid
In fact, in some organisms (most notably, the yeast Sac-
droplets, and CoQ levels ranged from 3-25% of normal
charomyces cerevisiae) Complex I is not even present!
Similarly, Complex V is absolutely required for oxida-
fibroblasts, and lymphoblast cell lines of affected
tive ATP synthesis and thus, cannot be bypassed.
patients, implying that this is a tissue specific disorder.
It is worth noting that the mtDNA-encoded subunits
To date, no known mutations responsible for defective
of complexes I and IV specify the essential catalytic
CoQ activity have been identified. However, identifi-
activities of the holoprotein (witness the absence of
cation of patients is extremely important, as symptoms
“nDNA-encoded” subunits in these complexes in
prokaryotes). While the biological importance of the
mtDNA-encoded “core” subunits is obvious, the rela-tive contribution of the nDNA-encoded subunits to
Discussion
overall function (e.g. modulation of activity commensu-
Although the first nuclear-encoded gene defect of the
rate with varying metabolic needs of the cell; tissue-spe-
respiratory chain was reported six years ago, the past
cific activity) is far less clear. As is the case with com-
two years have seen the identification of many more
plex V, however, it is possible that the functions of the
such errors. These mutations are not only in proteins
nDNA-encoded subunits of Complexes III and IV are as
that comprise the enzyme complexes themselves, but
essential as those of the mtDNA-encoded subunits, in
are also in “ancillary” proteins required for their assem-
which case mutations in nuclear genes would lead to
bly and proper functioning. With the sequencing of the
embryonic lethals. It is noteworthy in this regard that
entire human genome, the development of DNA chip
even the known assembly mutations in Complex IV in
arrays, and a greater understanding of the role that the
infants who at least survive birth do not obliterate COX
respiratory chain plays in the pathophysiology of human
enzyme activity completely, but rather allow affected
disease, it is predictable that many more nuclear muta-
tissues to maintain a residual (albeit low, and ultimately
tions will be identified in the near future.
insufficient) level of normal respiratory function. Of
In contrast to the diversity of phenotypic expression
course, mutations in “less critical” proteins within the
associated with mitochondrial DNA mutations, each
respiratory chain complexes may result in less deleteri-
mendelian-inherited mitochondrial disorder usually
ous effects or in subclinical disease, and may thus go
causes a distinct clinical phenotype, such as Leigh syn-
drome or, less commonly, fatal infantile cardioen-
Finally, mutations in Complex II in hereditary para-
cephalomyopathy. Also, while disorders due to mtDNA
ganglioma represent the first errors in a respiratory
mutations often have late onset, pathogenic mutations in
chain gene associated causally with neoplastic transfor-
nuclear genes controlling respiratory chain complexes
mation (4). While mutations in mitochondrial DNA
seem to cause diseases of infancy or early childhood.
have been postulated to play a role in tumorigenesis
This may be because recessive mendelian disorders tend
(39), no solid evidence supporting this concept had been
to be “all-or-none” phenotypically, in the sense that both
obtained prior to this finding. It is possible that abnor-
alleles must be mutated for the disease to be expressed.
mal respiratory chain function may play an important
In contrast, most mtDNA-based disorders are hetero-
role in the activation or inhibition of tumor suppressor
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
genes, but identification of other mutations and their
12. Fischer JC, Ruitenbeel W, Gabreels FJ, Janseen AJ,
pathophysiologic mechanisms will be necessary to con-
Renier WO, Sengers RCA, Stadhouders AM, ter Laak HJ,J.M.
encephalomyopathy: the first case with an establisheddefect at the level of coenzyme Q. Eur J Pediatr 144: 441-
Acknowledgements
This work was supported by grants from the Nation-
13. Haller RG, Henriksson KG, Jorfeld L, Hultman E, Wilbom
al Institutes of Health (NS11766, NS28828, NS39854,
R, Sahlin K, Areskog NH, Gunder M, Ayyad K, Blomqvist
and HD32062), the Muscular Dystrophy Association,
CG, Hall RE, Thuillier P, Kennaway NG, Lewis S (1991)Deficiency of skeletal muscle succinate dehydrogenase
and a Neil Hamilton Fairley NHMRC Postdoctoral Fel-
and aconitase. Pathophysiology of exercise in a novel
human muscle oxidative defect. J Clin Invest 88: 1197-1206
References
14. Jaksch M, Hofmann S, Kleinle S, Liechti-Gallati S, Pon-
Adams PL, Lightowlers RN, Turnbull DM (1997) Molecu-
gratz DE, Müller-Höcker J, Jedele KB, Meitinger T, Gerb-
lar analysis of cytochrome c oxidase deficiency in Leigh’s
itz KD (1998) A systemic mutation screen of 10 nuclear
and 25 mitochondrial candidate genes in 21 patients withcytochrome c
Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coul-
tRNA(Ser)(UCN) mutations in a subgroup with syndromal
son AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger
encephalopathy. J Med Genet 35: 895-900
F, Schreier PH, Smith AJH, Staden R, Young IG (1981)Sequence and organization of the human mitochondrial
15. Jaksch M, Ogilvie I, Yao J, Kortenhaus G, Bresser H-G,
Gerbitz K-D, Shoubridge EA (2000) Mutations in SCO2are associated with a distinct form of hypertrophic car-
Babcock GT, Wilkstrom M (1992) Oxygen activation and
diomyopathy and cytochrome c oxidase deficiency. Hum
the conservation of energy in cell respiration. Nature 356:
16. Keightley JA, Hoffbuhr KC, Burton MD, Salas VM, John-
Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC,
ston WSW, Penn AMW, Buist NRM, Kennaway NG (1996)
Myssiorek D, Bosch A, van der Mey A, Taschner PE,
A microdeletion in cytochrome c oxidase (COX) subunit III
Rubinstein WS, Myers EN, Richard CW, Cornelisse CJ,
associated with COX deficiency and recurrent myoglobin-
Devilee P, Devlin B (2000) Mutations in SDHD, a mito-
chondrial complex II gene, in hereditary paraganglioma. Science 287: 848-851
17. Kirby DM, Crawford M, Cleary MA, Dahl HH, Dennett X,
Thorburn DR (1999) Respiratory chain complex I defi-
Boitier E, Degoul F, Desguerre I, Charpentier C, Francois
ciency: an underdiagnosed energy generation disorder.
D, Ponsot G, Diry M, Rustin P, Marsac C (1998) A case of
mitochondrial encephalopathy associated with a musclecoenzyme Q10 deficiency. J Neurol Sci 156: 41-46
18. Loeffen J, Smeitink J, Triepels R, Smeets R, Schuelke M,
Sengers R, Trijbels F, Hamel B, Mullaart R, van den Heuv-
Bourgeron T, Rustin P, Chretien D, Birch-Machin M, Bour-
el L (1998) The first nuclear-encoded complex I mutation
geois M, Viegas-Pequignot E, Munnich A, Rötig A (1995)
in a patient with Leigh syndrome. Am J Hum Genet 63:
Mutation of a nuclear succinate dehydrogenase gene
results in mitochondrial respiratory chain deficiency. NatGenet 11: 144-149
19. Luft R, Ikkos D, Palmieri G, Ernster L, Afzelius B (1962) A
case of severe hypermetabolism of nonthyroid origin with
Burgeois M, Goutieres F, Chretien D, Rustin P, Munnich A,
a defect in the maintenance of mitochondrial respiratory
Aicardi J (1992) Deficiency in complex II of the respirato-
control: a correlated clinical, biochemical, and morpholog-
ry chain presenting as leukodystrophy in two sisters with
ical study. J Clin Invest 41: 1776-1804
20. Manfredi G, Schon EA, Moraes CT, Bonilla E, Berry GT,
Chandel NS, Maltepe E, Goldwasser E, Mathieu CE,
Sladky JT, DiMauro S (1995) A new mutation associated
Simon MC, Schumacker PT (1998) Mitochondrial reactive
with MELAS is located in a mitochondrial DNA polypep-
oxygen species trigger hypoxia-induced transcription.
tide-coding gene. Neuromusc Disord 5: 391-398
Proc Natl Acad Sci USA 95: 11715-11720
21. Matsuoka T, Maeda H, Goto Y-I, Nonaka I (1991) Muscle
Coenen MJH, van den Heuvel LP, Nijtmans LGJ, Morava
coenzyme Q10 in mitochondrial encephalomyopathies.
E, Marquardt I, Girschick HJ, Trijbels FJM, Grivell LA,
Smeitink JAM (1999) SURFEIT-1 gene analysis and two-dimensional blue native gel electrophoresis in cytochrome
22. Michel H, Behr J, Harrenga A, Kannt A (1998)
c oxidase deficiency. Biochem Biophys Res Commun 265:
Cytochrome c oxidase: structure and spectroscopy. AnnuRev Biophys Biomol Struc 27: 329-356
10. DiMauro S, Bonilla E, Lee CP, Schotland DL, Scarpa A,
23. Morris AAM, Leonard JV, Brown GK, Bidouki SK, Bindoff
Conn H, Chance B (1976) Luft’s disease: further bio-
LA, Woodward CE, Harding AE, Lake BD, Harding BN,
chemical and ultrastructural studies of skeletal muscle in
Farrell MA, Bell JE, Mirakhur M, Turnbull D (1996) Defi-
the second case. J Neuro Sci 27: 217-232
ciency in respiratory chain complex I is a common causeof Leigh disease. Ann Neurol 40: 25-30
11. DiMauro S (1999) Mitochondrial encephalomyopathies:
back to Mendelian genetics. Ann Neurol 45: 693-694
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
24. Ogasahara S, Yorifuji S, Nishikawa Y, Takahashi M, K. W,
38. Servidei S, Spinazzola A, Crocianin P, Ricci E, Silvestri G,
Hazama T, Nakamura Y, Hashimoto S, Kono N, Tarui S
Keller K, Simone P, DiMauro S (1996) Replacement ther-
(1985) Improvement of abnormal pyruvate metabolism
apy is effective in familial mitochondrial encephalomyopa-
and cardiac conduction defect with coenzyme Q10 in
thy with muscle coenzyme Q10 deficiency. Neurology 46:
Kearns-Sayre syndrome. Neurology 35: 372-77
25. Ogasahara S, Engel AG, Frens D, Mack D (1989) Muscle
39. Shay JW, Werbin H (1987) Are mitochondrial DNA muta-
coenzyme Q deficiency in familial mitochondrial
tions involved in the carcinogenic process? Mutat Res
encephalomyopathy. Proc Natl Acad Sci USA 86: 2379-
40. Smeitink J, van den Heuvel L (1999) Human mitochondr-
26. Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishi-
ial complex I in health and disease. Am J Hum Genet 64:
no I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum
DM, Van Coster R, Lyon G, Scalais E, Lebel R, Kaplan P,
41. Sobreira C, Hirano M, Shanske S, Keller RK, Haller RG,
Shanske S, De Vivo DC, Bonilla E, Hirano M, DiMauro S,
Davidson E, Santorelli FM, Miranda AF, Bonilla E, Mojon
Schon EA (1999) Fatal infantile cardioencephalomyopa-
DS, Barreira AA, King MP, DiMauro S (1997) Mitochondr-
thy with COX deficiency and mutations in SCO2, a COX
ial encephalopathy with coenzyme Q10 deficiency.
assembly gene. Nat Genet 23: 333-337
27. Parfait B, Chretien D, Rötig A, Marsac C, Munnich A,
42. Sue CM, Karadimas C, Checcarelli N, Tanji K,
Rustin P (2000) Compound heterozygous mutations in
Papadopoulou LC, Pallotti F, Guo FL, Shanske S, Hirano
the flavoprotein gene of the respiratory chain complex II in
M, De Vivo DC, van Coster R, Kaplan P, Bonilla E, DiMau-
a patient with Leigh syndrome. Hum Genet 106: 236-243
ro S (2000) Differential features of patients with mutations
28. Poyau A, Buchet K, Bouzidi MF, Zabot MT, Echenne B,
in two COX assembly genes, SURF-1 and SCO2. Ann
Yao J, Shoubridge EA, Godinot C (2000) Missense muta-
tions in SURF1 associated with deficient cytochrome c
43. Taylor RW, Birch-Machin MA, Schaefer J, Taylor L, Shakir
oxidase assembly in Leigh syndrome patients. Hum
R, Ackerell BAC, Cochran B, Bindoff LA, Jackson MJ,
Griffiths P, Turnbull DM (1996) Deficiency of complex II of
29. Rahman S, Blok R, Dahl H-HM, Danks DM, Kirkby DM,
the mitochondrial respiratory chain in late-onset optic
Chow CW, Christodoulou J, Thorburn DR (1996) Leigh
atrophy and ataxia. Ann Neurol 39: 224-232
syndrome: clinical features and biochemical and DNA
44. Teraoka M, Yokoyama Y, Ninomiya S, Inoue C, Yamashita
abnormalities. Ann Neurol 39: 343-351
S, Seino Y (1999) Two novel mutations of SURF1 in Leigh
30. Rivner MH, Shamsnia M, Swift TR, Trefz J, Roesel RA,
syndrome with cytochrome c oxidase deficiency. Hum
Carter AL, Yanamura W, Hommes FA (1989) Kearns-
Sayre syndrome and complex II deficiency. Neurology 39:
45. Tiranti V, Hoertnagel K, Carrozzo R, Galimberti C, Munaro
M, Granatiero M, Zelante L, Gasparini P, Marzella R, Roc-
31. Robinson BH, Glerum DM, Chow W, Petrova-Benedict R,
chi M, Bayona-Bafaluy MP, Enriquez JA, Uziel G, Bertini
Lightowlers R, Capaldi R (1990) The use of skin fibroblast
E, Dionisi-Vici C, Franco B, Meitinger T, Zeviani M (1998)
culture in the detection of respiratory chain defects in
Mutations of SURF-1 in Leigh disease associated with
patients with lacticacidemia. Pediatr Res 28: 549-555
cytochrome c oxidase deficiency. Am J Hum Genet 63:
32. Robinson BH (1998) Human complex I deficiency: clinical
spectrum and involvement of oxygen free radicals in the
46. Tiranti V, Jaksch M, Hofmann S, Galimberti C, Hoertnagel
pathogenicity of the defect. Biochim Biophys Acta 1364:
K, Lulli L, Freisinger P, Bindoff L, Gerbitz KD, Comi GP,
Uziel G, Zeviani M, Meitinger T (1999) Loss-of-function
33. Rustin P, Lebidois J, Chretien D, Bourgeron T, Piechaud
mutations of SURF-1 are specifically associated with
JF, Rötig A, Sidi D, Munnich A (1993) The investigation of
Leigh syndrome with cytochrome c oxidase deficiency.
respiratory chain disorders using endocardial biopsies. J
47. Tiranti V, Galimberti C, Nijtmans L, Bovolenta S, Perini
34. Saraste M (1999) Oxidative phosphorylation at the fin de
MP, Zeviani M (1999) Characterization of SURF-1 expres-
sion and Surf-1p function in normal and disease condi-tions. Hum Mol Genet 8: 2533-2540
35. Scarpulla RC (1997) Nuclear control of respiratory chain
expression in mammalian cells. J Bioenerget Biomemb
48. Triepels RH, van den Heuvel LP, Loeffen JLCM, Buskens
CAF, Smeets RJP, Gozalbo MFR, Budde SMS, MarimanEC, Wijburg FA, Barth PG, Trijbels JMF, Smeitink JAM
36. Schon EA, Bonilla E, DiMauro S (1997) Mitochondrial
(1999) Leigh syndrome associated with a mutation in the
DNA mutations and pathogenesis. J Bioenerg Biomembr
NDUFS7 (PSST) nuclear encoded subunit of complex I.
37. Schuelke M, Smeitink J, Mariman E, Loeffen J, Plecko B,
49. Valnot I, Kassis J, Chretien D, de Lonlay P, Parfait B,
Trijbels F, Stöckler-Ipsiroglu S, van den Heuvel L (1999)
Munnich A, Kachaner J, Rustin P, Rötig A (1999) A mito-
Mutant NDUFV1 subunit of mitochondrial complex I caus-
chondrial cytochrome b mutation but no mutations of
es leukodystrophy and myoclonic epilepsy. Nat Genet 21:
nuclearly encoded subunits in ubiquinol cytochrome c
reductase (complex III) deficiency. Hum Genet 104: 460-466
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
50. van den Heuvel L, Ruitenbeek W, Smeets R, Gelman-
Kohan Z, Elpeleg O, Loeffen J, Trijbels F, Mariman E, D. dB, Smeitink J (1998) Demonstration of a new pathogen-ic mutation in human complex I deficiency: a 5-bp dupli-cation in the nuclear gene encoding the 18-kD (AQDQ)subunit. Am J Hum Genet 62: 262-268
51. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G,
Mootha V, Troy A, Cinit C, Lowell B, Scarpulla RC,Spiegelman BM (1999) Mechanisms controlling mito-chondrial biogenesis and respiration through the thermo-genic coactivator-PGC-1. Cell 98: 115-124
52. Yamada EW, Huzel NJ (1992) Distribution of the ATPase
inhibitor proteins if mitochondria in mammalian tissuesincluding fibroblasts from a patient with Luft’s disease. Biochim Biophys Acta 1139: 143-147
53. Yao J, Shoubridge EA (1999) Expression and functional
analysis of SURF1 in Leigh syndrome patients withcytochrome c oxidase deficiency. Hum Mol Genet 8: 2541-2549
54. Zeviani M, Corona P, Nijtmans L, Tiranti V (2000) Nuclear
gene defects in mitochondrial disorders. Ital J Neurol Sci20: 401-408
55. Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, Cuth-
bert AP, Newbold RF, Wang J-c, Chevrette M, Brown GK,Brown RM, Shoubridge EA (1998) SURF1, encoding afactor involved in the biogenesis of cytochrome c oxidase,is mutated in Leigh syndrome. Nature Genet 20: 337-343
56. Zierz S, Jahns G, Jerusalem F (1989) Coenzyme Q in
serum and muscle of 5 patients with Kearns-Sayre syn-drome and 12 patients with ophthalmoplegia plus. J Neu-rol 236: 97-101
C. M. Sue and E. A. Schon: Mitochondrial Respiratory Chain Diseases and Mutations in Nuclear DNA
Europäisches Patentamt European Patent Office Office européen des brevets EP 0 989 848 B1 EUROPEAN PATENT SPECIFICATION (51) Int Cl.7: A61K 9/28 of the grant of the patent: 29.09.2004 Bulletin 2004/40 PCT/IB1998/000883 (21) Application number: 98921690.8 (22) Date of filing: 08.06.1998 WO 1998/056360 (17.12.1998 Gazette 1998/50) (54) FILM-COATED TABLET FOR IMP
References 1) Deutscher Bundestag Drucksache 16/12000, Bericht des Ausschusses für Bildung, Forschung und Technikfolgenabschätzung. Zukunftsreport Individualisierte Medizin und Gesundheitssystem (http://dip21.bundestag.de/dip21/btd/16/120/1612000.pdf) 2) Heimpel H, Wendt F. Congenital dyserythropoietic anemia with karyorrhexis and multinuclearity of erythroblasts. Helv Med Acta
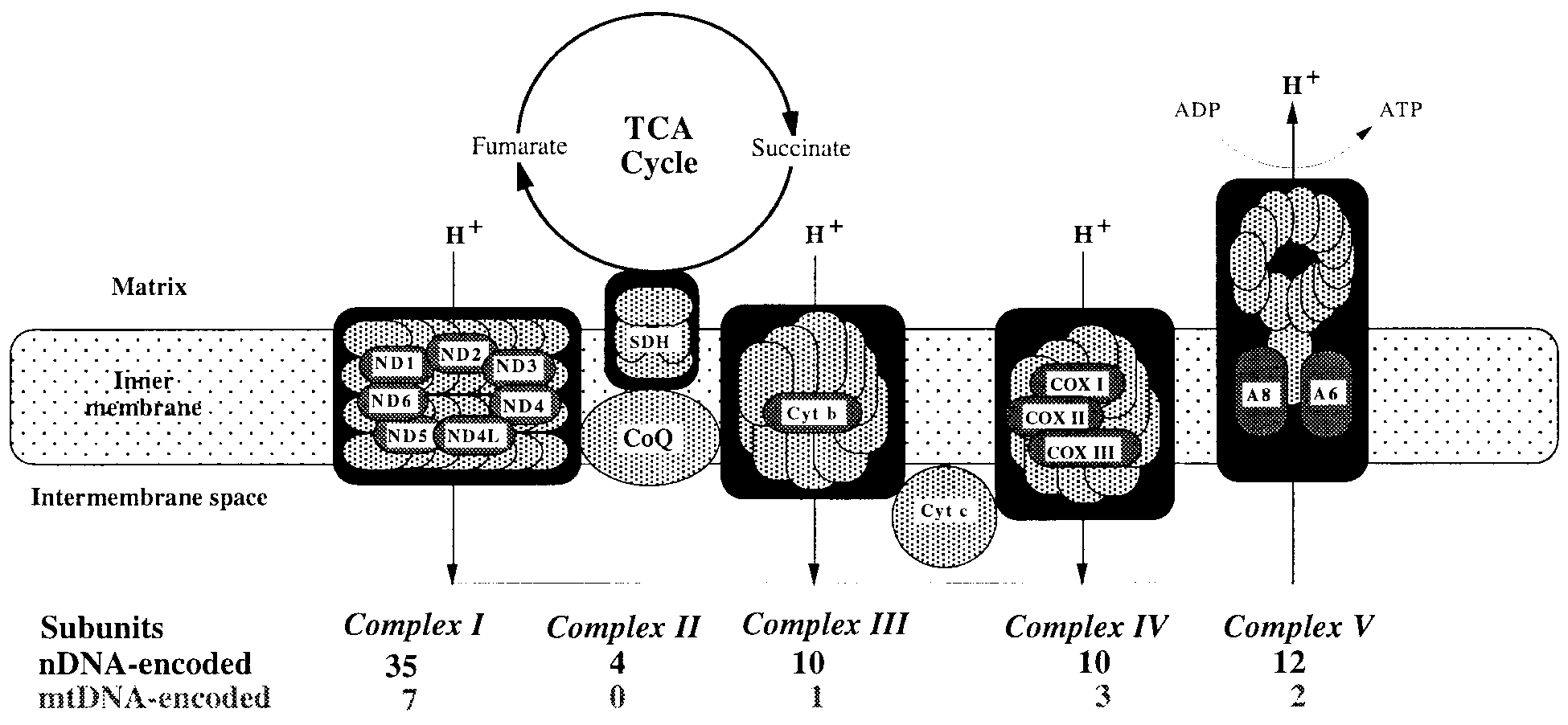 Figure 1. The mitochondrial respiratory chain, showing nDNA-encoded (light-shaded) and mtDNA-encoded (dark-shaded) subunits.
Figure 1. The mitochondrial respiratory chain, showing nDNA-encoded (light-shaded) and mtDNA-encoded (dark-shaded) subunits.